Co je fenylketonurie
Tam fenylketonurie (P.K.U.) jde o autozomálně recesivně dědičné metabolické onemocnění, které postihuje 1 z 10 000 jedinců a zdá se, že se vyskytuje více v homozygotnosti než v heterozygotech.
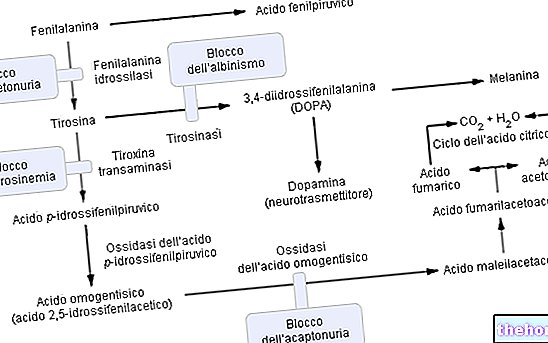
Fenylketonurie patřící do skupiny hyperfenylalaninémie významně narušuje metabolismus fenylalaninu a zejména jeho převod na tyrosin; fenylketonurie je rozpoznávána zvýšenými hladinami fenylalaninu a některých derivátů v moči (fenylpyruvát, fenylacetát, fenylaktát a fenylacetylglutamin).
Nejzávažnější komplikací fenylketonurie je mentální zpoždění.
Fenylalanin, tyrosin a deriváty
Fenylalanin je esenciální aminokyselina a tvoří většinu bílkovin v potravě; může být přeměněn enzymem fenylalaninhydroxyláza v tyrosinu (přidáním hydroxylové skupiny -OH). Na druhé straně je tyrosin prekurzorovou aminokyselinou pro syntézu:
- L-DOPA (meziprodukt syntézy dopaminu)
- Epinefrin
- Norepinefrin (všechny neurotransmitery).

Mechanismus fenylketonurie (P.K.U.)
Jak se očekávalo, u fenylketonurie je v důsledku jedné nebo více (celkem 6) chromozomálních mutací exprese (tedy metabolická aktivita) fenylalaninhydroxylázy prakticky nulová. Tyto změny mohou být různého druhu (od změn "missense" až po "sestřihové" defekty nebo dokonce "částečné delece"), ale důležité je, že kvůli této enzymatické neúčinnosti jsou hladiny fenylalaninu v krvi (obvykle 1 mg / 100 ml) v DOMINANTNÍ fenylketonurii snadno dosáhnou množství i 50krát vyššího.
Fungování enzymu fenylalaninhydroxylázy: K produkci tyrosinu (+ dihydrobiopterinu) vyžaduje fenylalaninhydroxyláza: fenylalanin, kyslík a tetrahydrobiopterin (redukovaný pteridin, který působí jako koofaktor); reakce je také reverzibilní a dihydrobiopterin lze znovu převést (díky enzymu dihydropterin reduktáza) v tetrahydrobiopterinu.
Komplikace
Fenylketonurie může na základě závažnosti patologického projevu a včasnosti diagnózy způsobit více či méně závažné komplikace; jako dědičná patologie se fenylketonurie rozlišuje v:
- Dominantní, proto charakterizovaný KOMPLETNÍ neaktivitou enzymu fenylalaninhydroxylázy
- Recesivní, ve kterém je aktivních pouze 30% celkového enzymatického dědictví.
Komplikace fenylketonurie lze přičíst, a přímo úměrné, metabolické akumulaci fenylalaninu, jeho derivátů a snížené syntéze tyrosinu. V patologii je přebytečný fenylalanin poměrně účinně filtrován ledvinami, které jej pouze částečně reabsorbují a vylučují močí ; nicméně přetrvávání hladin hyper-fenylalaninémie určuje metabolickou reakci molekulárního PŘEVODU v kyselina fenylpyruvová a / nebo jiné deriváty snadněji odčerpatelné (fenylpyruvát, fenylacetát, fenylaktát).
Fenylketonurie komplikuje toxicita fenylalaninu, kyseliny fenylpyruvové a jejích derivátů vůči centrálnímu nervovému systému (CNS). Jejich nadměrná přítomnost ve vývoji mozku neúprosně určuje formu mentální retardace.
Pozn. Plazmatické koncentrace ostatních aminokyselin jsou mírně sníženy, pravděpodobně kvůli zpětné vazbě na střevní absorpci nebo renální tubulární reabsorpci.
Poškození mozku, jako závažná komplikace fenylketonurie, je způsobeno odečtením jiných esenciálních aminokyselin v proteosyntéze, zejména při tvorbě polyribozomů, myelinu, noradrenalinu a serotoninu. Fenylketonurie - není viditelná bezprostředně po narození, ale po několika letech - pokud není léčena, vyžaduje hospitalizaci dítěte a je zcela nevratná.
Pokročilá fenylketonurie může být také jasně viditelná pouhým okem; vysoké koncentrace fenylalaninu, inhibující enzym tyrosinase, významně narušují syntézu melaninu snížením pigmentace kůže a vlasů; kromě toho akumulace fenylacetátu ve vlasech a pokožce dává fenylketonurikům silný a nepříjemný „myší pach“.












.jpg)