Všeobecnost
Cystická fibróza je nejčastějším autozomálně recesivním onemocněním v kavkazské populaci, které postihuje přibližně 1 z 2 500 jedinců.
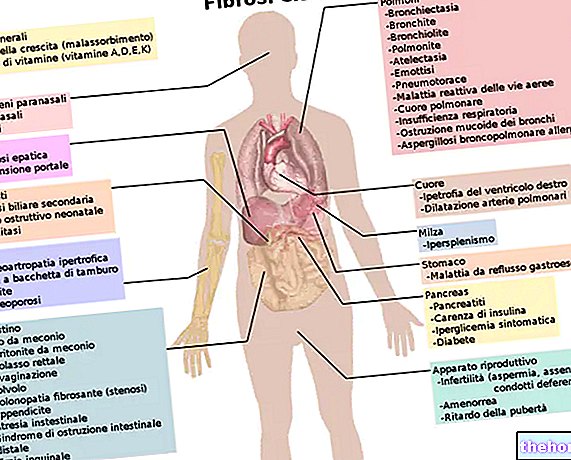
Tento patologický stav je známý svými škodlivými účinky na dýchací systém, ale ovlivňuje také jiné systémy, jako je trávicí a reprodukční systém.
U jedinců s cystickou fibrózou jsou dýchací cesty ucpané hustým a viskózním hlenem, který je obtížné vyčistit i při nejsilnějším kašli. Dýchání se stává obtížným a pacienti - pokud se nevyvíjejí neustálé snahy udržovat dýchací cesty čisté několikrát denně - riskují, že zemřou na vlastní sekreci. Pacienti s cystickou fibrózou často umírají na zápal plic, protože ucpané dýchací cesty poskytují úrodné prostředí pro růst bakterií.

Příčiny
Cystická fibróza je způsobena mutacemi v genu pro regulátor transmembránové vodivosti (CFTR) cystické fibrózy umístěného na chromozomu 7 (mapování lokusu: 7q31).
Je známo nejméně 1 500 mutací genu CFTR. Nejčastější mutace se běžně nazývá „Delta-F508“ (DF508) a je způsobena delecí 3 párů bází v exonu 10, což má za následek ztrátu fenylalaninu v poloze 508.
Protein kódovaný genem CFTR je transmembránový kanál patřící do superrodiny transportních ATPáz nebo ABC transportérů, umístěný na úrovni apikální membrány epiteliálních buněk a zodpovědný za transport chlorového iontu.
Za normálních podmínek určité buňky lemující dýchací cesty vylučují hlen spolu s vodnou kapalinou, která snižuje jeho hustotu. Při cystické fibróze je sekrece vodné tekutiny značně snížena, v důsledku čehož se hlen stává velmi hustým a obtížně odstranitelným z dýchacího traktu.
V respiračním epitelu, stejně jako u všech epitelů nesoucích kapalinu, závisí transport vody na transportu rozpuštěných látek. Buňky respiračního epitelu k vylučování vody aktivně transportují ionty chloru (Cl-) z intersticiální tekutiny do lumen a vytvářejí negativní elektrický potenciál, který způsobuje pasivní tok sodíku (Na +) ve stejném směru. Na + a Cl - zvyšují osmotický tlak kapaliny, která zvlhčuje stranu epitelu obrácenou k lumenu, v důsledku čehož se voda pohybuje pasivně podle osmotického gradientu, od intersticiální kapaliny k lumenu. Genová vada ovlivňující nástup cystická fibróza brání transportu Cl- přímo a nepřímo interferuje s transportem Na + a vody.V důsledku toho se v epitelu nevytváří osmotický gradient nezbytný pro sekreci vody.
Rizikové faktory
- Rodinné dědictví. Vzhledem k tomu, že cystická fibróza je dědičné onemocnění, které se přenáší autozomálně recesivním způsobem, je důležité vzít v úvahu rodinnou anamnézu (anamnézu) budoucích rodičů.
Pokud tedy děti zdědí pouze jednu kopii (pouze jednoho nemocného rodiče), nevyvine se u nich cystická fibróza, ale budou asymptomatickými nosiči a potenciálně by mohly defektní gen předat svým dětem. Jak je znázorněno na obrázku, když dva zdravé nosiče (heterozygotní pro gen CFTR, tedy nesoucí pouze jednu kopii abnormálních genů) mají dítě, existuje šance jedna ke čtyřem (25%), že dítě je postiženo cystickou fibrózou ( homozygotní pro gen CFTR).
- Populace sounáležitosti. Výskyt cystické fibrózy je vyšší u lidí severního a evropského původu.

Klinické příznaky a příznaky
Další informace: Příznaky cystické fibrózy
Závažnost příznaků se může lišit v závislosti na průběhu onemocnění: většina klinických příznaků postihuje dýchací a gastrointestinální systém.