Aktivní složky: Paricalcitol
Zemplar 2 mikrogramy Měkké tobolky
Příbalové letáky Zemplar jsou k dispozici pro velikosti balení:- Zemplar 1 mikrogram měkké tobolky
- Zemplar 2 mikrogramy Měkké tobolky
- Zemplar 5 mikrogramů / ml injekční roztok
Proč se používá Zemplar? K čemu to je?
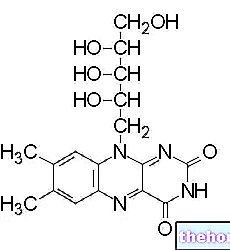
Zemplar je syntetická forma aktivního vitaminu D.
Vitamín D ve své aktivní formě zajišťuje normální funkci mnoha tkání v našem těle, včetně příštítných tělísek a kostí. U lidí s normální funkcí ledvin je tato aktivní forma vitaminu D přirozeně produkována ledvinami, ale v případě selhání ledvin je produkce aktivního vitaminu D značně snížena. Zemplar proto poskytuje zdroj aktivního vitaminu D, když tělo není schopné produkovat dostatečné množství, a pomáhá předcházet následkům nízkých hladin aktivního vitaminu D u pacientů s renální insuficiencí (fáze 3, 4 a 5), tj. Vysokými hladinami parathormonu, které mohou způsobit problémy s kostmi.
Kontraindikace Kdy by neměl být Zemplar používán
Neužívejte Zemplar
- jestliže jste alergický / á (přecitlivělý / á) na paricalcitol nebo na kteroukoli další složku přípravku Zemplar (uvedenou v bodě 6).
- jestliže máte v krvi vysoké hladiny vápníku nebo vitaminu D.
Váš lékař vás bude moci informovat, pokud váš případ spadá do výše uvedených dvou podmínek.
Opatření pro použití Co potřebujete vědět před užitím přípravku Zemplar
- Před zahájením léčby je důležité omezit množství fosforu ve vaší stravě.
- Ke kontrole hladin fosforu mohou být potřebná pojiva fosforu. Pokud užíváte vázače fosforu na bázi vápníku, měl by vám lékař upravit dávku.
- Váš lékař nařídí několik krevních testů, aby sledoval vaši léčbu.
- U některých pacientů s chronickým onemocněním ledvin stupně 3 a 4 byly pozorovány zvýšené hladiny látky zvané kreatinin. Toto zvýšení se však neprojevuje snížením funkce ledvin.
Interakce Které léky nebo potraviny mohou změnit účinek přípravku Zemplar
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval (a) nebo které možná budete užívat.
Některé léky mohou ovlivnit způsob účinku přípravku Zemplar nebo zvýšit pravděpodobnost výskytu nežádoucích účinků. Je zvláště důležité informovat svého lékaře, pokud užíváte ketokonazol (používá se k léčbě plísňových infekcí, jako je kandidóza nebo drozd), cholestyramin (používá se ke snížení hladiny cholesterolu) jestliže užíváte léky na srdce nebo krevní tlak (například digoxin a diuretika nebo pilulky na odstranění přebytečné vody z našeho těla) nebo léky s vysokým obsahem vápníku. Je také důležité si uvědomit, že užíváte léky obsahující hořčík nebo hliník, například některá antacida a pojiva fosforu.
Před užitím jakýchkoli jiných léků se poraďte se svým lékařem nebo lékárníkem.
Zemplar s jídlem a pitím
Zemplar lze užívat s jídlem nebo mezi jídly.
Varování Je důležité vědět, že:
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat. Potenciální riziko proto není známé, proto by měl být parikalcitol používán pouze v nezbytně nutných případech.
Není známo, zda se parikalcitol vylučuje do lidského mléka. Pokud užíváte přípravek Zemplar, poraďte se před kojením se svým lékařem.
Před užitím jakýchkoli jiných léků se poraďte se svým lékařem nebo lékárníkem.
Řízení dopravních prostředků a obsluha strojů
Zdá se, že Zemplar neovlivňuje schopnost řídit nebo obsluhovat stroje.
Zemplar obsahuje ethanol
Tento léčivý přípravek obsahuje malé množství ethanolu (alkoholu), méně než 100 mg v tobolce, které může upravit nebo zvýšit účinek jiných léků. To může způsobit újmu osobám s onemocněním jater, alkoholismem, epilepsií, kteří utrpěli poškození mozku nebo trpí nemocemi, stejně jako těhotné nebo kojící ženy a děti.
Dávka, způsob a doba podání Jak používat Zemplar: Dávkování
Vždy užívejte přípravek Zemplar přesně podle pokynů svého lékaře. Pokud máte pochybnosti, poraďte se se svým lékařem nebo lékárníkem.
Chronické selhání ledvin Fáze 3 a 4
Obvyklá dávka je jedna tobolka denně nebo obden, až třikrát týdně. Na základě výsledků laboratorních testů vám lékař určí vhodnou dávku. Jakmile je léčba přípravkem Zemplar zahájena, je pravděpodobné, že bude provedena úprava dávky v závislosti na tom, jak na léčbu reagujete. Váš lékař vám pomůže určit správnou dávku přípravku Zemplar.
Chronické selhání ledvin Fáze 5
Obvyklá dávka je jedna tobolka denně nebo obden, až třikrát týdně. Na základě výsledků laboratorních testů vám lékař určí vhodnou dávku. Jakmile je léčba přípravkem Zemplar zahájena, je pravděpodobné, že bude provedena úprava dávky v závislosti na tom, jak na léčbu reagujete. Váš lékař vám pomůže určit správnou dávku přípravku Zemplar.
Transplantace ledvin
Obvyklá dávka je jedna tobolka denně nebo obden až třikrát týdně. Na základě výsledků laboratorních testů vám lékař určí vhodnou dávku. Jakmile je léčba přípravkem Zemplar zahájena, je pravděpodobné, že bude provedena úprava dávky v závislosti na tom, jak na léčbu reagujete. Váš lékař vám pomůže určit správnou dávku přípravku Zemplar.
Onemocnění jater
Pokud máte mírné nebo středně závažné onemocnění jater, není nutná úprava dávky. U pacientů s těžkým onemocněním jater však nejsou žádné zkušenosti.
Použití u dětí a dospívajících
Neexistují žádné informace o používání tobolek Zemplar u dětí.
Použití u starších osob
Existují „omezené zkušenosti“ s používáním přípravku Zemplar u pacientů starších 65 let. Obecně nebyly mezi pacienty ve věku 65 let nebo staršími a mladšími pacienty pozorovány žádné rozdíly v účinnosti a bezpečnosti léčiva.
Jestliže jste zapomněl (a) užít Zemplar:
Pokud zapomenete užít dávku svého léku, vezměte si ji okamžitě, jakmile si vzpomenete. Pokud je však již čas na další dávku, neužívejte vynechanou dávku; jednodušeji, pokračujte v užívání Zemplaru podle pokynů svého lékaře (dávka a čas).
Nezdvojnásobujte následující dávku, abyste nahradil (a) vynechanou dávku.
Jestliže jste přestal (a) užívat přípravek Zemplar:
Je důležité pokračovat v užívání přípravku Zemplar podle pokynů svého lékaře, pokud jste nebyli výslovně instruováni, abyste jej přestali užívat.
Máte -li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Předávkování Co dělat, když jste užil příliš mnoho přípravku Zemplar
Předávkování přípravkem Zemplar může způsobit abnormální zvýšení hladin vápníku v krvi, což může být škodlivé. Mezi příznaky, které se mohou objevit brzy po předávkování přípravkem Zemplar, patří pocit slabosti a / nebo necitlivosti, bolest hlavy, nevolnost (pocit na zvracení) nebo zvracení, sucho v ústech, zácpa, bolest svalů nebo kostí a kovová chuť.
Mezi příznaky, které se mohou vyskytnout po delší dobu užívání příliš velkého množství přípravku Zemplar, patří: ztráta chuti k jídlu, ospalost, ztráta hmotnosti, oční potíže, rýma, svědění, pocit horka a horečky, ztráta libida, silná bolest břicha (v důsledku zánětu slinivka břišní) a ledvinové kameny.Krevní tlak se může změnit a může se objevit nepravidelný srdeční tep (palpitace). Výsledky testů krve a moči mohou ukázat zvýšený cholesterol, močovinu a dusík a zvýšení hladin jaterních enzymů. Zemplar může zřídka způsobit mentální změny včetně zmatenosti, ospalosti , nespavost nebo podrážděnost.
Pokud užijete příliš mnoho přípravku Zemplar nebo si všimnete některého z výše uvedených příznaků, okamžitě se poraďte s lékařem.
Nežádoucí účinky Jaké jsou vedlejší účinky přípravku Zemplar
Podobně jako všechny léky, může mít i Zemplar nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Okamžitě informujte svého lékaře, pokud zaznamenáte některý z následujících nežádoucích účinků:
U pacientů s chronickým selháním ledvin stadia 3 a 4
Mezi nejběžnější účinky (nejméně 1 ze 100 pacientů) patří vyrážka a bolest žaludku.
Může dojít ke zvýšení krevních hladin látky zvané vápník, jakož i látky zvané fosforečnan vápenatý, která je odvozena od množství vápníku k množství jiné látky v krvi zvané fosfát (u pacientů s významným chronickým onemocněním ledvin) .
Méně časté účinky (nejméně 1 z 1000 pacientů) jsou alergické reakce (jako jsou potíže s dýcháním, dušnost, vyrážka, svědění nebo otok obličeje a rtů), svědění kůže a kopřivka, zácpa, sucho v ústech, svalové křeče, závratě a změny chuti . Rovněž mohou být změněny testy jaterních funkcí.
Pokud se u vás objeví alergická reakce, okamžitě kontaktujte svého lékaře.
U pacientů s chronickým selháním ledvin stupeň 5
Nejčastějšími nežádoucími účinky (nejméně 1 ze 100 pacientů) jsou průjem, pálení žáhy (reflux nebo poruchy trávení), snížená chuť k jídlu, závratě, bolest prsou a akné.Mohou se také objevit změny hladiny vápníku v krvi.
Nejčastějšími vedlejšími účinky (nejméně 1 ze 100 pacientů) pozorovanými u pacientů během intravenózního užívání parikalcitolu jsou: bolest hlavy, poruchy chuti, svědění, snížení hladiny parathormonu, zvýšení hladiny vápníku a zvýšení hladiny fosforu.
Méně časté nežádoucí účinky (nejméně 1 z 1 000 pacientů) pozorované u pacientů užívajících intravenózní parikalcitol jsou: nepravidelný srdeční tep, prodloužená doba krvácení, abnormální funkce jaterních testů, ztráta hmotnosti, zástava srdečního tepu, tachyarytmie, snížený počet bílých krvinek, snížený počet červených krvinek počet, zvětšené žlázy, mrtvice, přechodný ischemický záchvat, kóma, mdloby, závratě, záškuby, brnění, necitlivost, zvýšený oční tlak, mírně zarudlé oči, červené oči, bolest ucha, plicní edém, krvácení z nosu, dušnost, sípání, kašel, mírné střevní krvácení anální krvácení, bolest žaludku, potíže s polykáním, syndrom dráždivého tračníku, průjem, zácpa, pálení žáhy, zvracení, nevolnost, sucho v ústech, nepříjemné pocity v žaludku, svědivá vyrážka, vyrážka, puchýře, vypadávání vlasů, růst vlasů li, noční pocení, bolest v místě vpichu, pocit pálení kůže, bolest kloubů, bolest svalů, ztuhlost kloubů, bolest zad, svalové záškuby, zvýšené hladiny parathormonu v krvi, ztráta chuti k jídlu, snížená chuť k jídlu, krevní infekce, infekce krve, zápal plic, chřipka, nachlazení, bolest v krku, vaginální infekce, rakovina prsu, nízký krevní tlak (hypotenze), vysoký krevní tlak (hypertenze), bolest na hrudi, neobvyklá chůze, oteklé nohy, otoky, nepříjemné pocity na hrudi, horečka, slabost, bolest, únava, malátnost žízeň, pocit nepohodlí, bolest prsou, alergie, potíže s erekcí, poruchy vědomí, zmatenost, úzkost, nespavost, podrážděnost, agitovanost.
Hlášení nežádoucích účinků
Pokud se u vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Nežádoucí účinky můžete hlásit také přímo na adresu: Státní ústav pro kontrolu léčiv Šrobárova 48100 41 Praha 10 Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
Expirace a retence
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nepoužívejte Zemplar po uplynutí doby použitelnosti, uvedené na krabičce a štítku za EXP. Datum exspirace se vztahuje k poslednímu dni uvedeného měsíce.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Pomůže to chránit životní prostředí.
Jiná informace
Co Zemplar obsahuje
- Léčivou látkou je paricalcitol. Jedna měkká tobolka obsahuje 2 mikrogramy parikalcitolu.
- Dalšími složkami jsou: triglyceridy se středním řetězcem, ethanol, butylhydroxytoluen.
- Obal tobolky obsahuje: želatinu, glycerol, vodu, oxid titaničitý (E171), černý oxid železitý (E172) a žlutý oxid železitý (E172).
- Tisková barva obsahuje: propylenglykol, černý oxid železitý (E172), polyvinylacetátftalát, Macrogol 400, hydroxid amonný.
Jak Zemplar vypadá a obsah balení
Zemplar Soft Capsules, 2 mikrogramy, je oranžová, oválná měkká tobolka označená logem a iniciálami ZF.
Každé balení obsahuje 1 nebo 4 blistry. Každý blistr obsahuje 7 tobolek.
Zdroj příbalové informace: AIFA (Italská agentura pro léčivé přípravky). Obsah zveřejněný v lednu 2016. Přítomné informace nemusí být aktuální.
Abyste měli přístup k nejaktuálnější verzi, doporučujeme navštívit webovou stránku AIFA (Italská agentura pro léčivé přípravky). Prohlášení a užitečné informace.
01.0 NÁZEV LÉČIVÉHO PŘÍPRAVKU
MĚKKÉ Kapsle ZEMPLAR
02.0 KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tobolka přípravku Zemplar 1 mikrogram obsahuje 1 mikrogram parikalcitolu.
Jedna tobolka přípravku Zemplar 2 mcg obsahuje 2 mcg paricalcitolu.
Pomocná látka se známými účinky:
Jedna tobolka přípravku Zemplar 1 mikrogram obsahuje 0,71 mg ethanolu.
Jedna tobolka přípravku Zemplar 2 mcg obsahuje 1,42 mg ethanolu.
Úplný seznam pomocných látek viz bod 6.1.
03.0 LÉKOVÁ FORMA
Měkké tobolky
1 mikrogramová tobolka: šedá, oválná měkká tobolka označená ZA
2 mcg tobolka: oranžově hnědá, oválná měkká tobolka označená iniciálami ZF
04.0 KLINICKÉ INFORMACE
04.1 Terapeutické indikace
Zemplar je indikován k prevenci a léčbě sekundární hyperparatyreózy u dospělých pacientů s chronickým selháním ledvin (fáze 3 a 4) a chronickým selháním ledvin v konečném stadiu (stupeň 5) podstupujícím hemodialýzu nebo peritoneální dialýzu.
04.2 Dávkování a způsob podání
Dávkování
Chronické selhání ledvin (CKD), fáze 3 a 4
Zemplar by měl být podáván jednou denně nebo třikrát týdně každý druhý den.
Počáteční dávkování
Počáteční dávka by měla být vypočítána s ohledem na základní hladiny intaktního parathormonu (iPTH).
Úprava dávky
Dávka by měla být individualizována, tj. Stanovena individuálně na základě sérových nebo plazmatických hladin iPTH, sledováním sérového vápníku a sérových fosfatémií. Tabulka 2 nabízí příklad doporučeného postupu pro úpravu dávky.
Po zahájení léčby a během období úpravy dávky je třeba pečlivě sledovat hladiny vápníku v séru. Pokud je u pacienta podstupující terapii vazači fosforu na bázi vápníku pozorována „hyperkalcémie nebo trvale zvýšený produkt fosforečnanu vápenatého vyšší než 55 mg2 / dl2 (4,4 mmol2 / l2), dávka nebo přerušení podávání. Případně podání přípravku Zemplar by měla být snížena nebo dočasně přerušena.Pokud je léčba ukončena, podávání léku by mělo být obnoveno nižší dávkou, kdy dojde k normalizaci produktu vápníku a fosforečnanu vápenatého.
Fáze 5 Chronické selhání ledvin (CKD)
Zemplar se podává třikrát týdně, každý druhý den.
Počáteční dávkování
Počáteční dávka přípravku Zemplar v mcg by měla být vypočtena z výchozích hladin intaktních parathormonů = iPTH (pg / ml) / 60 [(pmol / l) / 7] až do maximální počáteční dávky 32 mcg.
Úprava dávky
Dávka by měla být individualizována, tj. Individuálně stanovena, a měla by být založena na sérových hladinách intaktního parathormonu, vápníku a fosforu. Doporučená úprava dávky kapslí parikalcitolu je založena na následujícím vzorci:
Úprava dávky
nebo
Úprava dávky
Po zahájení terapie, během období úpravy dávky a ve spojení s podáváním silných inhibitorů P450 3A je třeba pečlivě sledovat hladiny vápníku a fosforu. Pokud je zaznamenána hyperkalcémie nebo zvýšený produkt kalcium xfosfor a pokud pacient podstupuje terapii vazači fosforu na bázi vápníku, měla by být jeho dávka snížena nebo podávání přerušeno. Alternativně může pacient přejít na pojivo fosforu, které není na bázi vápníku.
Pokud je vápník> 11,0 mg / dl (2,8 mmol / l) nebo produkt Ca x P> 70 mg2 / dl2 (5,6 mmol2 / l2) nebo iPTH ≤ 150 pg / ml, dávku je třeba snížit o 2 - 4 mcg z toho vypočítaného na základě nejnovější hladiny iPTH / 60 (pg / ml) [iPTH / 7 (pmol / l)]. V případě, že je nutná další úprava dávky, podávání kapslí parikalcitolu by mělo být sníženo nebo ukončeno, dokud se tyto parametry normalizují.
Když se hladina iPTH blíží referenčnímu rozmezí (150-300 pg / ml), může být zapotřebí malé individuální úpravy dávky, aby se dosáhlo stabilní úrovně iPTH. Monitorování hladin iPTH, vápníku nebo fosforu lze provádět méně často než jednou týdně, lze použít mírnější počáteční dávku / poměr úpravy dávky.
Zvláštní populace
Porucha funkce jater:
U pacientů s mírnou až středně těžkou poruchou funkce jater není nutná úprava dávky.
U pacientů s těžkou poruchou funkce jater nejsou žádné zkušenosti (viz bod 5.2).
Transplantace ledvin:
Pacienti s transplantací ledvin s chronickým selháním ledvin stadia 3 a 4 a sekundární hyperparatyreózou nebyli během klinických studií fáze 3 studováni. Na základě publikovaných studií byl u pacientů, kteří podstoupili transplantaci ledviny s chronickým renálním stadiem 3 a 4, na základě publikovaných studií uveden počáteční dávka a algoritmus úpravy dávky Selhání a sekundární hyperparatyreóza jsou stejné jako u pacientů s jednoduchým chronickým selháním ledvin stupně 3 a 4 a sekundární hyperparatyreózou. Sérové hladiny vápníku a fosforu by měly být pečlivě sledovány po zahájení léčby, během období úpravy dávky a při současném podávání silných inhibitory cytochromu P450 3A.
Pediatrická populace:
Bezpečnost a účinnost přípravku Zemplar Capsule u dětí do 18 let nebyla dosud stanovena.
Aktuálně dostupné údaje jsou popsány v bodě 5.1, ale nelze učinit žádná doporučení ohledně dávkování.
Senioři:
Mezi staršími pacienty (65 - 75 let) a mladšími pacienty nebyly pozorovány žádné celkové rozdíly v bezpečnosti a účinnosti léčiva, ale nelze vyloučit možnost, že někteří starší jedinci jsou citlivější.
Způsob podání
Zemplar lze užívat s jídlem nebo bez jídla.
04.3 Kontraindikace
Parikalcitol by neměl být předepisován pacientům s prokázanou toxicitou vitaminu D, hyperkalcémií nebo přecitlivělostí na parikalcitol nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
04.4 Zvláštní upozornění a vhodná opatření pro použití
Nadměrné potlačení sekrece parathormonu může vést ke zvýšeným hladinám vápníku v séru a může vést k nízkému obratu kostních chorob. Za účelem získání adekvátních fyziologických referenčních hodnot by mělo být provedeno pečlivé sledování pacienta a individuální titrace dávky.
Pokud se vyvine klinicky významná „hyperkalcémie“ a pacient je na terapii vázačem fosforu na bázi vápníku, je třeba dávku tohoto chelátoru snížit nebo podávání přerušit.
Chronická hyperkalcémie může být spojena s generalizovanou kalcifikací cév a jinými kalcifikacemi měkkých tkání.
Léčivé přípravky obsahující fosfát nebo vitamín D by neměly být užívány současně s parikalcitolem kvůli zvýšenému riziku hyperkalcémie a nárůstu produktu Ca x P (viz bod 4.5).
Digitalisem indukovaná toxicita je potencována přítomností jakékoli příčiny hyperkalcémie, proto je třeba při předepisování digitalisu souběžně s parikalcitolem postupovat velmi opatrně (viz bod 4.5).
U pacientů na dialýze může parikalcitol, stejně jako jiné aktivátory receptorů vitaminu D, způsobit zvýšení sérového kreatininu (a následně snížit průměrnou rychlost glomerulární filtrace GFR [eGFR]), aniž by došlo ke změně skutečné rychlosti glomerulární filtrace (GFR).
Je -li parikalcitol podáván současně s ketokonazolem, je třeba postupovat velmi opatrně (viz bod 4.5).
Zvláštní upozornění na pomocné látky:
Tento léčivý přípravek obsahuje malé množství ethanolu (alkoholu), méně než 100 mg na 1 kapsli 1 mcg a 2 mcg. Toto množství může být škodlivé u subjektů trpících alkoholismem (viz body 2 a 4.2). Je třeba vzít v úvahu u těhotných nebo kojících žen, dětí a vysoce rizikových skupin, jako jsou pacienti s onemocněním jater nebo epilepsií.
04.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Ketokonazol: O ketokonazolu je známo, že je nespecifickým inhibitorem různých enzymů cytochromu P450. Dostupné údaje in vivo a in vitro naznačují, že ketokonazol může interagovat s enzymy odpovědnými za metabolismus parikalcitolu a dalších analogů vitaminu D. Při podávání parikalcitolu současně s ketokonazolem je třeba postupovat velmi opatrně. Účinek opakovaných dávek ketokonazolu podávaných v dávkách 200 mg dvakrát denně (BID) po dobu 5 dnů na farmakokinetiku parikalcitolových tobolek byl studován u zdravých subjektů. V přítomnosti ketokonazolu byla Cmax parikalcitolu ovlivněna jen zanedbatelně, ale AUC0- ¥ se téměř zdvojnásobil. Průměrný poločas parikalcitolu byl 17,0 hodin v přítomnosti ketokonazolu, ve srovnání s 9,8hodinovým poločasem, kdy byl parikalcitol podáván samostatně (viz bod 4.4). Výsledky této studie naznačují, že po orálním nebo intravenózním podání parikalcitolu je nepravděpodobné, že by maximální zvětšení AUCINF parikalcitolu v důsledku lékové interakce s ketokonazolem bylo větší než dvojnásobek.
Nebyly provedeny žádné specifické studie interakcí. Toxicita vyvolaná digitalisem je zvýšena hyperkalcémií způsobenou jakoukoli příčinou, proto je třeba postupovat s maximální opatrností v případě pacientů podstupujících léčbu parikalcitolem, kteří musí současně užívat digitalis.
Fosfát nebo léčivé přípravky související s vitamínem D by neměly být užívány současně s parikalcitolem, protože může existovat zvýšené riziko hyperkalcémie a může dojít ke zvýšení produkce Ca x P (viz bod 4.4).
Vysoké dávky kalciových přípravků nebo thiazidových diuretik mohou zvýšit riziko hyperkalcémie.
Přípravky obsahující hořčík (např. Antacida) by neměly být užívány současně s přípravky vitaminu D, protože může dojít k hypermagnezémii.
Hliníkové přípravky (např. Antacidy, chelátory fosforu) by neměly být podávány současně s přípravky s vitamínem D při chronické terapii, protože může dojít ke zvýšení hladiny hliníku v krvi a může dojít k toxicitě kostí z hliníku.
Léky, které snižují střevní absorpci vitamínů rozpustných v tucích, jako je cholestyramin, mohou interferovat s absorpcí kapslí Zemplar.
04.6 Těhotenství a kojení
Těhotenství
Adekvátní údaje o podávání parikalcitolu těhotným ženám nejsou k dispozici. Studie na zvířatech odhalily reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známo. Parikalcitol by proto neměl být během těhotenství používán, pokud to není nezbytně nutné.
Čas krmení
Není známo, zda se parikalcitol vylučuje do lidského mléka. Studie na zvířatech ukázaly, že parikalcitol nebo jeho metabolity jsou v malém množství vylučovány do mateřského mléka. Rozhodnutí pokračovat nebo přerušit kojení nebo pokračovat nebo přerušit léčbu Zemplarem by mělo být zváženo s ohledem na přínos kojení pro kojence a přínos terapie Zemplarem pro matku.
04.7 Účinky na schopnost řídit a obsluhovat stroje
Zemplar má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
04.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Bezpečnost kapslí parikalcitolu byla hodnocena ve třech multicentrických, dvojitě zaslepených, placebem kontrolovaných, 24týdenních klinických studiích zahrnujících 220 pacientů s chronickým selháním ledvin, fáze 3 a 4 a v multicentrické klinické studii. placebem kontrolovaná studie zahrnující 88 pacientů s chronickým selháním ledvin, fáze 5. Kromě toho jsou ze dvou dalších studií k dispozici údaje o postmarketingových zkušenostech s kapslemi paricalcitolu. Nejčastěji hlášenými nežádoucími účinky u pacientů užívajících parikalcitol jsou hyperkalcémie a zvýšený produkt fosforečnanu vápenatého. V klinických studiích fáze 3/4 a stupně 5 byla incidence hyperkalcémie Zemplar (3/167, 2%) oproti placebu (0/137, 0%) a zvýšeným produktem fosforečnanu vápenatého byl Zemplar (19/167, 11 %) oproti placebu (8/137, 6%).
Tabulkový seznam nežádoucích účinků
Všechny nežádoucí účinky související s měkkými tobolkami Zemplar jsou uvedeny v tabulce 3 podle konvencí orgánových systémů MedDRA a podle frekvence. Frekvence jsou definovány následovně: velmi časté (≥1 / 10), časté (≥1 / 100,
Tabulka 3: Nežádoucí reakce pozorované u měkkých tobolek Zemplar v klinických studiích a ze zkušeností po uvedení přípravku na trh.
* Četnost nežádoucích účinků vyplývajících ze zkušeností po uvedení přípravku na trh nelze odhadnout a byla hlášena jako „Není známo“.
† Tato nežádoucí reakce byla pozorována ve studiích u pacientů před dialýzou (viz také bod 4.4).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky, ke kterým dochází po registraci léčivého přípravku, je důležité, protože umožňuje průběžné sledování poměru přínosů a rizik léčivého přípravku. Zdravotničtí pracovníci jsou požádáni, aby hlásili jakékoli podezření na nežádoucí účinky prostřednictvím národního systému hlášení. "Adresa www. agenziafarmaco.gov.it/it/responsabili.
04.9 Předávkování
Nadměrné podávání kapslí Zemplar může způsobit hyperkalcémii, hyperkalciurii, hyperfosfatémii a „nadměrné potlačení parathormonu. Vysoký příjem vápníku a fosfátů souběžně s kapslemi Zemplar může vyvolat podobné změny“.
Léčba pacientů s „klinicky významnou hyperkalcémií spočívá v okamžitém snížení dávky nebo přerušení léčby parikalcitolem a zahrnuje zavedení diety s nízkým obsahem vápníku, pozastavení podávání“ doplňků obsahujících vápník, mobilizaci pacientů, sledování nerovnováhy elektrolytů a tekutin, vyhodnocení změn v elektrokardiografické stopě (kritické u pacientů podstupujících terapii digitalisem) a „hemodialýzou nebo peritoneální dialýzou s dialyzátem bez vápníku, na základě toho, co je považováno za vhodné.
Příznaky a příznaky intoxikace vitamínem D spojené s hyperkalcémií zahrnují:
Časné příznaky a příznaky: astenie, bolest hlavy, ospalost, nevolnost, zvracení, sucho v ústech, zácpa, myalgie, bolest kostí, kovová chuť.
Pozdní příznaky a symptomy: anorexie, ztráta hmotnosti, kalcifikovaná konjunktivitida, pankreatitida, fotofobie, rhinorea, pruritus, hypertermie, snížené libido, zvýšení dusíku močoviny v krvi, hypercholesterolemie, zvýšené transaminázy, ektopická kalcifikace, hypertenze, srdeční arytmie, ospalost, smrt a zřídka zjevná psychóza.
Hladiny vápníku v séru by měly být často monitorovány, dokud se normalizují.
Parikalcitol není významně eliminován dialýzou.
05.0 FARMAKOLOGICKÉ VLASTNOSTI
05.1 Farmakodynamické vlastnosti
Farmakoterapeutická kategorie: antiparatyroidní látky.
ATC kód: H05BX02.
Akční mechanismus
Paricalcitol je syntetický analog kalcitriolu, biologicky aktivní formy vitaminu D, s modifikacemi na postranním řetězci (D2) a na kruhu A (19-nor). Na rozdíl od kalcitriolu je paricalcitol selektivním aktivátorem vitaminu D (VDR Parikalcitol selektivně stimuluje receptory vitaminu D v příštítných tělískách, aniž by způsobil zvýšení receptorů vitaminu D ve střevě a je méně aktivní při resorpci kostí. Parikalcitol dále stimuluje receptory citlivé na vápník (CaSR) přítomné v příštítných tělískách. V důsledku toho parikalcitol snižuje hladiny parathormonu (PTH) inhibicí proliferace příštítných tělísek a snížením syntézy a sekrece PHT, s minimálním dopadem na hladiny vápníku a fosforu; paricalcitol může působit přímo na osteoblasty, aby zachoval objem kosti a zlepšil mineralizační povrchy. Oprava změněných hladin parathormonu spolu s normalizací homeostázy vápníku a fosforu může zabránit nebo vyléčit metabolické kostní onemocnění spojené s chronickým selháním ledvin.
Klinická účinnost
Fáze 3 a 4 chronického selhání ledvin
Primárního koncového ukazatele účinnosti léčiva alespoň dvou po sobě jdoucích snížení o ≥ 30% oproti výchozímu iPTH dosáhlo 91% pacientů léčených tobolkami parikalcitolu a 13% pacientů léčených placebem (kostní alkalická pphosphatase - sérově specifická a sérový osteokalcin byla významně snížena ( renální dysfunkce, odhadovaná rychlost glomerulární filtrace (podle vzorce MDRD) a sérový kreatinin u pacientů léčených parikalcitolovými tobolkami ve srovnání s pacienty léčenými placebem. U významně většího počtu pacientů léčených parikalcitolovými kapslemi došlo ke snížení proteinurie, jak bylo zjištěno měření provedená semikvantitativní metodou (měrkou) ve srovnání s pacienty léčenými placebem.
Fáze 5 Chronické selhání ledvin
Primárního cílového parametru účinnosti léčiva alespoň dvou po sobě následujících snížení o ≥ 30% oproti výchozímu iPTH dosáhlo 88% pacientů léčených parikalcitolovými tobolkami a 13% pacientů léčených placebem (p
Klinické údaje shromážděné u pediatrických pacientů po podání injekčního roztoku Zemplar (intravenózního):
Bezpečnost a účinnost injekčního roztoku Zemplar byla zkoumána v randomizované, dvojitě zaslepené, placebem kontrolované studii s 29 pediatrickými pacienty ve věku 5 až 19 let s konečným stadiem chronického selhání ledvin podstupujících hemodialýzu. Šest nejmladších pacientů léčených roztokem Zemplar pro injekci ve studii byli ve věku od 5 do 12 let. Počáteční dávka injekčního roztoku Zemplar byla 0,04 mcg / kg 3krát týdně, pokud byla výchozí intaktní parathormon (iPTH)
05.2 Farmakokinetické vlastnosti
Vstřebávání
Paricalcitol se dobře vstřebává. U zdravých subjektů byla po perorálním podání parikalcitolu v hodnotě 0,24 mcg / kg průměrná absolutní biologická dostupnost přibližně 72%; maximální plazmatická koncentrace (Cmax) se rovnala 0,630 ng / ml (1,512 pmol / ml) po 3 hodinách a plocha pod křivkou koncentrace v čase (AUC0- ¥) se rovnala 5,25 ng • h / ml (12,60 pmol • h Průměrná absolutní biologická dostupnost u pacientů podstupujících hemodialýzu a peritoneální dialýzu byla 79%, respektive 86%, s horní hranicí 95% intervalu spolehlivosti rovnající se 93%, respektive 112%. Studie interakce s potravinami provedená na zdravé subjekty uvedly, že Cmax a „AUC0-? zůstávají nezměněny, pokud je parikalcitol podáván současně s jídlem s vysokým obsahem tuku ve srovnání s podáváním nalačno. Zemplar Capsule lze proto užívat také mezi jídly.
Cmax a AUC0-a parikalcitolu se zvyšovaly úměrně v rozmezí dávek 0,06 až 0,48 mcg / kg u zdravých subjektů. Po opakovaných dávkách bylo expozice v ustáleném stavu dosaženo do sedmi dnů u zdravých subjektů, které užívaly lék denně nebo třikrát týdně.
Rozdělení
Parikalcitol se ve velké míře váže na plazmatické bílkoviny (> 99%).Poměr koncentrace parikalcitolu v krvi k plazmatické koncentraci parikalcitolu byl v průměru 0,54 v rozmezí koncentrací 0,01 až 10 ng / ml (0,024 až 24 pmol / ml), což naznačuje, že buňky S krví bylo spojeno velmi malé množství léčiva. Průměrný zdánlivý objem distribuce po podání dávky 0,24 mcg / kg parikalcitolu u zdravých subjektů byla 34 litrů.
Biotransformace
Po orálním podání dávky 0,48 μg / kg 3H-parikalcitolu bylo původní léčivo rozsáhle metabolizováno a pouze asi 2% vyloučené dávky byly získány neporušené ve stolici, zatímco žádná nebyla detekována v moči. . Asi 70% radioaktivity bylo vyloučeno stolicí a 18% bylo získáno močí. Většina systémové expozice je způsobena původním léčivem. V lidské plazmě byly identifikovány dva menší metabolity parikalcitolu. Jeden metabolit byl identifikován jako 24 (R) -hydroxyparikalcitol, zatímco druhý metabolit nebyl identifikován. 24 (R) -hydroxyparikalcitol je méně účinný než parikalcitol v krysím modelu in vivo potlačení parathormonu.
Data in vitro naznačují, že parikalcitol je metabolizován různými jaterními a nehepatálními enzymy, včetně mitochondriálních CYP24, CYP3A4 a „UGT1A4. Mezi identifikované metabolity patří produkt 24 (R) -hydroxylace, stejně jako 24,26- a 24,28 -dehydroxylace a přímá glukuronidace.
Odstranění
U zdravých subjektů je průměrný poločas eliminace parikalcitolu pět až sedm hodin ve studovaném rozmezí dávek 0,06 až 0,48 mcg / kg. Stupeň akumulace byl v souladu s poločasem a frekvencí dávek. Hemodialýza nemá v podstatě žádný vliv na eliminaci parikalcitolu.
Zvláštní populace
Senioři
Farmakokinetika parikalcitolu nebyla studována u pacientů starších 65 let.
Pediatrie
Farmakokinetika parikalcitolu nebyla zkoumána u pacientů mladších 18 let.
Typ
Farmakokinetika parikalcitolu po podání jednotlivých dávek léčiva v rozmezí dávek 0,06 až 0,48 mcg / kg byla nezávislá na pohlaví.
Jaterní nedostatečnost
Ve studii provedené s intravenózním podáním Zemplaru byla porovnávána dostupnost parikalcitolu (0,24 mcg / kg) u pacientů s lehkou (n = 5) a středně těžkou (n = 5) poruchou funkce jater (podle metody Child-Pugh) a subjekty s normální funkcí jater (n = 10). Farmakokinetika nevázaného parikalcitolu byla podobná v celém rozsahu jaterních funkcí hodnocených v této studii. U pacientů s lehkou nebo středně těžkou poruchou funkce jater není nutná úprava dávky. Vliv na farmakokinetiku parikalcitolu za přítomnosti nedostatečnosti nebylo hodnoceno závažné onemocnění jater .
Selhání ledvin
Farmakokinetika parikalcitolu po podání jednotlivé dávky byla hodnocena u pacientů s chronickým selháním ledvin stupně 3 nebo středně těžkou poruchou funkce ledvin (n = 15, GFR = 36,9 - 59,1 ml / min / 1,73 m2), chronickým selháním ledvin stupně 4 nebo těžkou poruchou funkce ledvin ( n = 14, GFR = 13,1 - 29,4 ml / min / 1,73 m2) a chronické selhání ledvin stupeň 5 nebo konečné stadium onemocnění ledvin [n = 14 při hemodialýze (HD) a n = 8 při peritoneální dialýze (PD)]. Podobně jako u endogenního 1,25 (OH) 2 D3 byla farmakokinetika parikalcitolu po orálním podání významně ovlivněna přítomností renální insuficience, jak ukazuje tabulka 4. Ve srovnání se zdravými subjekty postihli pacienti chronické selhání ledvin fáze 3, 4, a 5 ukázaly pokles CL / F a prodloužení poločasu.
Tabulka 4. Srovnání průměrných ± SD farmakokinetických parametrů u pacientů s renálním selháním v různých fázích úcta zdravým subjektům
Po perorálním podání kapslí parikalcitolu byl farmakokinetický profil parikalcitolu u chronického selhání ledvin ve fázích 3–5 srovnatelný. Proto nejsou nutné žádné zvláštní úpravy dávky, kromě těch, které jsou výslovně doporučeny (viz bod 4.2).
05.3 Předklinické údaje vztahující se k bezpečnosti
Nejvýraznější nálezy ze studií toxicity po opakovaném podávání u hlodavců a psů jsou obecně připisovány kalcemické aktivitě parikalcitolu. Účinky, které jasně nesouvisely s hyperkalcemií, zahrnovaly snížení počtu bílých krvinek. U psů výskyt atrofie brzlíku u psů a přítomnost změněných hodnot aktivovaného částečného tromboplastinového času (zvýšené u psů, snížené u potkanů) .Bílé krvinky.
Bylo pozorováno, že parikalcitol nepříznivě neovlivňuje plodnost potkanů a že neexistuje žádný důkaz teratogenní aktivity u potkanů ani králíků. Vysoké dávky jiných přípravků vitaminu D podávané během březosti u zvířat vyvolaly teratogenezi. Bylo prokázáno, že paricalcitol nepříznivě ovlivňuje životaschopnost plodu a podporuje významné zvýšení perinatální a postnatální mortality novorozených potkanů, pokud je podáván v dávkách, u nichž bylo zjištěno, že jsou toxické pro matku.
Během série testů genetické toxicity in vitro a in vivoBylo prokázáno, že parikalcitol nevykazuje žádnou potenciální genetickou toxicitu.
Studie karcinogenity u hlodavců nenaznačují zvláštní rizika pro lidské použití.
Podávané dávky a / nebo systémové expozice parikalcitolu byly o něco vyšší než terapeutické dávky / systémové expozice (viz bod 4.2).
06.0 FARMACEUTICKÉ INFORMACE
06.1 Pomocné látky
Obsah kapslí:
Triglyceridy se středním řetězcem
Ethanol
Butylhydroxytoluen
Obal tobolky:
Černý inkoust:
Propylenglykol
Černý oxid železitý (E172)
Polyvinylacetátftalát
Macrogol 400
Hydroxid amonný
06.2 Neslučitelnost
Irelevantní.
06.3 Doba platnosti
2 roky.
06.4 Zvláštní opatření pro skladování
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
06.5 Charakter vnitřního obalu a obsah balení
Lahve z polyetylenu s vysokou hustotou (HDPE) s dětským bezpečnostním uzávěrem z polypropylenu. Každá lahvička obsahuje 30 tobolek.
Blistry z PVC / fluoropolymeru / hliníkové fólie obsahující 7 tobolek. Každé balení obsahuje 1 nebo 4 blistry balené v krabičkách obsahujících 7 nebo 28 tobolek.
Na trhu nemusí být všechny velikosti balení.
06.6 Návod k použití a zacházení
Žádné zvláštní pokyny.
07.0 DRŽITEL ROZHODNUTÍ O REGISTRACI
AbbVie S.r.l.
S.R.148 Pontina km 52 snc
04011 Campoverde di Aprilia (LT)
08.0 REGISTRAČNÍ ČÍSLO
Zemplar "1 Mcg Soft Capsules" 30 kapslí v lahvi Hdpe - AIC č. 036374039
Zemplar "1 Mcg Soft Capsules" 7 kapslí v blistru Pvc / fluoropolymer / Al - AIC č. 036374041
Zemplar "1 Mcg Soft Capsules" 28 kapslí v blistru Pvc / fluoropolymer / Al - AIC č. 036374054
Zemplar "2 Mcg Soft Capsules" 30 kapslí v lahvi Hdpe - AIC č. 036374066
Zemplar "2 Mcg Soft Capsules" 7 kapslí v blistru Pvc / fluoropolymer / Al - AIC č. 036374078
Zemplar "2 Mcg Soft Capsules" 28 kapslí v blistru Pvc / fluoropolymer / Al - AIC č. 036374080
09.0 DATUM PRVNÍ REGISTRACE NEBO PRODLOUŽENÍ REGISTRACE
Datum první registrace: 18. června 2009
10.0 DATUM REVIZE TEXTU
09/2016