Všeobecnost
Termín retinitis pigmentosa (RP) identifikuje skupinu genetických onemocnění charakterizovaných progresivní degenerací sítnice.

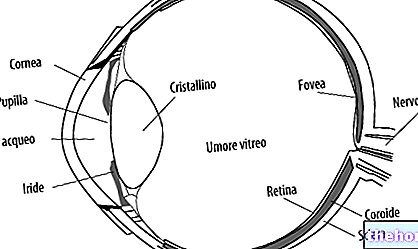
Retinitis pigmentosa je retinální dystrofie charakterizovaná postupnou ztrátou fotoreceptorů a dysfunkcí pigmentového epitelu, což znamená, že sítnice postupně snižuje svoji schopnost přenášet zrakové informace do mozku optickým nervem.
Patologický proces začíná změnami pigmentového epitelu sítnice. Jak postupuje retinitis pigmentosa, dochází ke ztenčení krevních cév zásobujících sítnici, které procházejí atrofií. Po vyšetření očního pozadí jsou vizuálně zjistitelné charakteristické depozity. Sítnicový pigment ( odtud název nemoci). Atrofické změny a poškození mohou také zahrnovat zrakový nerv a postupně odumírají fotosenzitivní buňky sítnice.
Pacienti postižení retinitis pigmentosa zpočátku pociťují problémy se zrakem zejména ve špatně osvětleném prostředí a stěžují si na zúžení periferního zorného pole. Centrální vidění je ušetřeno až do pozdějších fází onemocnění a konečný výsledek se může dramaticky lišit: mnoho lidí s retinitis pigmentosa si po celý život zachovává omezené vidění, zatímco jiní úplně ztrácejí zrak.
Retinitis pigmentosa je dědičné onemocnění, způsobené především genetickými změnami přenesenými od jednoho nebo obou rodičů. Typ genetické vady určuje, které sítnicové buňky se na poruše nejvíce podílejí, a umožňuje z klinického hlediska odlišit různé podmínky. K dnešnímu dni bylo identifikováno více než 50 různých genetických vad spojených s retinitis pigmentosa. Abnormality mohou být přeneseny z rodičů na potomky prostřednictvím jednoho ze tří dědičných vzorců: autozomálně recesivní, autozomálně dominantní nebo heterosomálně recesivně (X-spojený nebo X-spojený).
Příznaky
Další informace: Retinitis Pigmentosa Symptomy
Retinitis pigmentosa se obvykle vyskytuje u dospívajících a mladých dospělých. Příznaky se často objevují ve věku od 10 do 30 let, ale diagnózu lze stanovit v raném dětství nebo mnohem později v životě.
Počáteční příznaky retinitis pigmentosa mohou zahrnovat:
- Obtížné vidění v noci (noční slepota) nebo za špatných světelných podmínek
- Pomalé přizpůsobování od vidění ve tmě k vidění ve světle a naopak;
- Zúžení zorného pole a ztráta periferního vidění;
- Citlivost na světlo a oslnění.
Některé příznaky závisí na typu zapojených fotoreceptorů. Tyče jsou zodpovědné za černé a bílé vidění, zatímco kužely vám umožňují rozlišit barvy.
Ve většině případů retinitis pigmentosa jsou nejprve zapojeny tyčinky. V rychle se vyvíjejících formách však mohou být kužely postiženy také v rané fázi.
Tyčinky jsou soustředěny ve vnějších částech sítnice a jsou aktivovány tlumeným světlem, takže jejich degenerace ovlivňuje periferní a noční vidění. Pokud jsou zahrnuty kužely, je možné zažít ztrátu vnímání barev a centrálního vidění.
Převaha zapojených fotoreceptorů je dána konkrétní vadou přítomnou v pacientově genetickém složení.
Prvním příznakem retinitis pigmentosa je často noční slepota (nebo noktalopie). Někteří lidé zjišťují, že potřebují stále více času na přizpůsobení se rozdílům ve světle při přechodu z dobře osvětlené oblasti do tmavší. Typická forma ztráty zraku vyvolává zúžení periferního vidění (tunelové nebo teleskopické vidění); tomuto vzoru se říká prstencový skotom. Někdy tento jev v počátečních fázích chybí, ale je zaznamenán, když jedinec často zakopává o předměty nebo se účastní dopravní nehody. Když ztráta zraku zahrnuje centrální oblast sítnice (také nazývanou makulární dystrofie) mají potíže se čtením a podrobnou prací, která vyžaduje soustředění na jeden předmět, například provlékání nitě okem jehly Mnoho pacientů uvádí, že vidí záblesky světla (fotopsie), často popisované jako malá, blikající a blikající světla.
Rychlost progrese onemocnění a stupeň ztráty zraku se liší od člověka k člověku. Některé extrémní případy se mohou rychle vyvinout do dvou desetiletí, jiné pomalý kurz, který nikdy nevede k úplné slepotě. Časný nástup se vyskytuje u závažnějších forem retinitis pigmentosa, zatímco u pacientů s mírnějšími stavy (např. Autozomálně dominantní) může dojít k rozvoji onemocnění v páté nebo šesté dekádě života. V rodinách s X retinitis pigmentosa spojenou s X jsou muži postiženi častěji než ženy a vážněji; ženy na druhé straně přenášejí genetickou charakteristiku (nesou pozměněný gen na chromozomu X) a projevují příznaky poruchy méně často.
Komplikace
Retinitis pigmentosa bude i nadále postupovat, i když pomalu. Úplná slepota je však vzácná, ale může dojít k významnému snížení periferního a centrálního vidění.
U pacientů s retinitis pigmentosa se v útlém věku často vyvine otok sítnice (makulární edém) nebo šedý zákal. Tyto komplikace lze léčit, pokud narušují vidění.
Související nemoci
Běžně pacient s retinitis pigmentosa nemá žádné jiné poruchy a v tomto případě mluvíme o „nesyndromické“ nebo jednoduché retinitis pigmentosa. Několik syndromů však sdílí některé klinické příznaky s tímto očním onemocněním; nejčastější je Usherův syndrom, který postihuje přibližně 10-30% všech pacientů s retinitis pigmentosa a je spojen se souběžnou vrozenou nebo progresivní ztrátou sluchu. U Leberovy vrozené amaurózy však mohou děti během prvních šesti měsíců života oslepnout nebo téměř oslepnout. Mezi další nemoci související s retinitis pigmentosa patří Bardet-Biedlův syndrom a Refsumova choroba.
Příčiny
Onemocnění může být způsobeno řadou genetických vad: ve skutečnosti existuje několik genů, které, pokud jsou ovlivněny změnou, mohou způsobit fenotyp retinitis pigmentosa. Ty obvykle kódují proteiny zapojené do transdukční kaskády, která umožňuje vidění, faktory transkripce buněk (které vysílají chybné zprávy do sítnicových buněk) nebo pro prvky tvořící strukturu fotoreceptorů. Zděděné genové mutace jsou v buňkách přítomny od okamžiku početí; mezi běžné abnormality patří mutace genů RP1 (u retinitis pigmentosa-1, autozomálně dominantní) , RHO (RP4, autozomálně dominantní) a RDS (RP7, autozomálně dominantní). Dědičné příčiny retinitis pigmentosa jsou vzácné, ale možnost nalezení izolovaného případu (spontánní mutace), u kterého není přítomna rodinná anamnéza nemoc.

.jpg)