Všeobecnost
Osteogenesis imperfecta je vrozené genetické onemocnění, které není spojeno s pohlavím, je zodpovědné za určitou křehkost kostí a výrazný sklon ke zlomeninám.
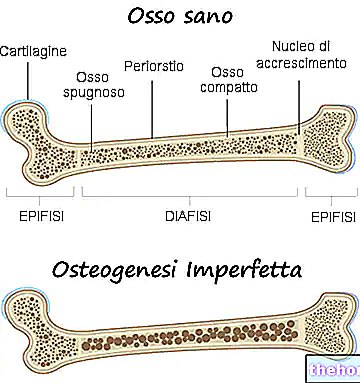
Příznaky osteogenesis imperfecta jsou četné; obecně se skládají z: oslabení kostí, vysokého sklonu ke zlomeninám kostí, přítomnosti modrých, šedých nebo purpurových očních skléer, přítomnosti kostních deformit nebo jiných změn skeletu, trojúhelníkového obličeje, křehkosti zubů atd. .
Pro správnou diagnózu osteogenesis imperfecta jsou obecně nezbytné následující: fyzikální vyšetření, anamnéza, lékařské zobrazovací testy, test na hodnocení kolagenu typu I a genetický test.
V současné době je bohužel jediná léčba dostupná pacientům s osteogenesis imperfecta symptomatická. Daná nemoc je ve skutečnosti nevyléčitelná.
Co je osteogenesis imperfecta?
Osteogenesis imperfecta je genetické onemocnění, které činí kosti postiženého slabší a náchylnější ke zlomeninám.
Ve skutečnosti lékaři termínem osteogenesis imperfecta označují heterogenní skupinu genetických chorob, charakterizovaných určitým stupněm křehkosti kostí. Existuje tedy několik forem (nebo typů) osteogenesis imperfecta, některé mnohem závažnější než jiné.
Je to vrozená nemoc
U lidí, kteří jsou touto nemocí postiženi, je osteogenesis imperfecta onemocnění přítomné od narození, a proto může být v každém smyslu definováno jako vrozené onemocnění.
SOUVISEJÍ SE SEX?
Osteogenesis imperfecta není geneticky podmíněné genetické onemocnění, jako je hemofilie nebo Klinefelterův syndrom.
EPIDEMILOGIE
Podle některých statistických výzkumů by se výskyt osteogenesis imperfecta rovnal jednomu případu každých 15 000–20 000 porodů. To znamená, že každých 15 000–20 000 novorozenců má jednoho postiženého osteogenesis imperfecta.
Jiné statistické studie také ukázaly, že osteogenesis imperfecta postihuje muže i ženy stejně a že nemá žádnou preferenci pro konkrétní populaci nebo etnickou skupinu.
Délka života je extrémně variabilní parametr, který závisí na formě osteogenesis imperfecta.
Příčiny
Osteogenesis imperfecta je téměř vždy výsledkem kvalitativní a kvantitativní změny produkce kolagenu typu I.
Kolagen typu I je nezbytný pro posílení kostí a pro udržení zdravých pojivových tkání tvořících chrupavky, šlachy, kůži, oční skléru atd.
Proto změna produkce kolagenu typu I ovlivňuje pevnost kostí a dobré zdraví pojivových tkání přítomných v lidském těle.
CO MĚNÍ VÝROBU KOLAGENU?
Genetická nemoc je stav, který vzniká v důsledku mutace jednoho nebo více genů tvořících buněčnou DNA.
V případě osteogenesis imperfecta lze jejich příčiny téměř vždy hledat v mutaci jednoho nebo obou genů COL1A1 (umístěných na chromozomu 17) a COL1A2 (umístěných na chromozomu 7).
Za normálních podmínek regulují COL1A1 a COL1A2 normální produkci kolagenu typu I; v přítomnosti mutací v jejich náboji selhávají ve své regulační funkci.
Důležité: jaké další geny, pokud jsou mutované, způsobují osteogenesis imperfecta?
Kromě mutací COL1A1 a COL1A2 jsou potenciálními příčinami osteogenesis imperfecta mutace v genech IFITM5, SERPINF1, CRTAP a LEPRE1.
Výše uvedené geny pokrývají funkce odlišné od COL1A1 a COL1A2 - proto nekontrolují produkci kolagenu typu I - ale stále mají „vliv na pevnost a odolnost kostí lidské kostry.
JAKÝ JE TO GENETICKÝ NEMOC?
Osteogenesis imperfecta je autozomálně genetické onemocnění.
Termín autosomální, spojený s genetickým onemocněním, naznačuje, že daný stav je způsoben genetickými mutacemi založenými na autozomálních a nesexuálních chromozomech.
Čtenářům se připomíná, že lidská bytost vlastní chromozomální soubor 23 párů celkových chromozomů, z nichž 22 párů je autozomálního typu a pouze jeden pár je sexuálního typu. Pár chromozomů sexuálního typu ovlivňuje pohlaví individuální.
Osteogenesis imperfecta po mutacích v COL1A1, COL1A2 a IFITM5 má všechny vlastnosti autozomálně dominantní choroby. Je -li způsobena mutacemi v genech SERPINF1, CRTAP a LEPRE1, má vlastnosti autosomálně recesivního onemocnění.
TYPY
V současné době se lékaři domnívají, že existuje 8 typů (nebo forem) osteogenesis imperfecta. Aby rozlišili různé typy, rozhodli se použít římské číslování, přesněji řečeno prvních osm římských číslic.
Níže uvedená tabulka ukazuje 8 forem osteogenesis imperfecta, mutace, které je způsobují, a další charakteristiky.
Chlap
Mutovaný gen
Typ genetického onemocnění
THE
COL1A1
Autozomálně dominantní
II
COL1A1 a COL1A2
Autozomálně dominantní
III
COL1A1 a COL1A2
Autozomálně dominantní
IV
COL1A1 a COL1A2
Autozomálně dominantní
PROTI.
IFITM5
Autozomálně dominantní
VY
SERPINF1
Autozomálně recesivní
VII
CRTAP
Autozomálně recesivní
VIII
ZÍSKAT 1
Autozomálně recesivní
* Pozn .: mutace v COL1A1 a COL1A2, které způsobují první čtyři formy osteogenesis imperfecta, jsou genetické změny s mírně odlišnými charakteristikami. Jinak by nemělo smysl rozlišovat jedno od druhého.
Příznaky, příznaky a komplikace
Všechny typy osteogenesis imperfecta jsou zodpovědné za oslabení kostí, takže osoba postižená nemocí má zvláštní predispozici ke zlomeninám. Stupeň oslabení kostí se liší podle tvaru; u některých z nich je toto oslabení větší než u jiných.
Když to bylo řečeno, je třeba zdůraznit, že každá forma osteogenesis imperfecta má svůj vlastní symptomatický obraz, který pro někoho může připomínat symptomatologický obraz jiných forem.
MOŽNÉ PŘÍZNAKY A ZNAKY
Mezi možné příznaky a příznaky osteogenesis imperfecta patří:
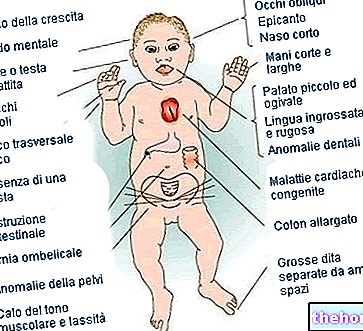
- Přítomnost kostních malformací;
- Přítomnost krátkého a malého těla (zamýšleno jako kufr);
- Problémy s klouby (např: uvolněné klouby);
- Svalová slabost;
- Modré, fialové nebo šedé oční skléry;
- Trojúhelníkový obličej;
- Sudová hruď;
- Morfologické anomálie páteře;
- Zubní křehkost;
- Pokles nebo úplná ztráta sluchu;
- Dýchací problémy
- Problémy související s absencí nebo nedostatkem kolagenu typu 1.
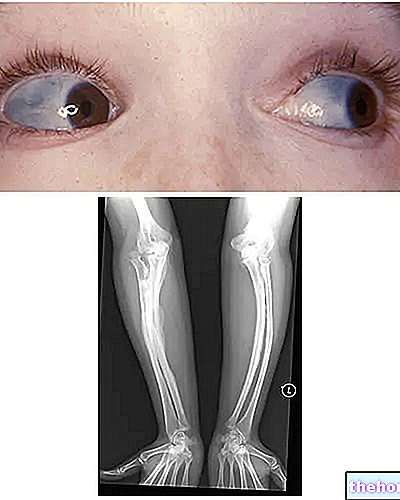
Osteogenesis Imperfecta: všimněte si modrého zbarvení sklerů a deformací kostí, které charakterizují onemocnění. Z wikipedia.org
JAKÉ JSOU NEJVÁŽNĚJŠÍ FORMY NEDOSTATEČNÉ OSTEOGENEZY?
Lékaři klasifikují symptomatologickou závažnost různých typů osteogenesis imperfecta na stupnici 3 stupňů, kterými jsou: mírný stupeň, střední stupeň a závažný stupeň.
Pouze jedna forma patří do kategorie „mírného stupně“: „osteogenesis imperfecta typu I“; 4 formy osteogenesis imperfecta patří do kategorie „mírného stupně“: IV, V a VI; konečně do kategorie „závažného stupně“ patří 3 formy: II, III, VII a VIII.
TYP I: VLASTNOSTI
Nejběžnější a nejméně závažná forma osteogenesis imperfecta typu I má následující vlastnosti:
- Způsobuje zlomeniny zejména před pubertou;
- Nemá „téměř žádný vliv na výšku, takže pacienti mají obvykle normální“ výšku;
- Způsobuje problémy s klouby a svalovou slabost
- Je zodpovědný za modré, fialové nebo šedé skléry;
- Je příčinou trojúhelníkových anomálií obličeje a páteře;
- Skoro nikdy nezpůsobuje kostní deformity. Pokud je to provokuje, jsou minimální;
- Může způsobit křehkost zubů a / nebo ztrátu sluchu (k tomu druhému obvykle dochází v dospělosti);
- Je spojena s přítomností kolagenu typu I, který má normální kvalitu, ale abnormální množství (je chudší než obvykle).
TYP II: VLASTNOSTI
Osteogenesis imperfecta typu II se vyznačuje:
- Příčina smrti při narození nebo krátce poté. Respirační problémy téměř vždy způsobují smrt;
- Přítomnost značné křehkosti kostí a závažných kostních deformit;
- Nízkého vzrůstu a nedostatečně vyvinutých plic
- Modré, purpurové nebo šedě zbarvené skléry;
- Přítomnost kvantitativních a kvalitativních anomálií kolagenu typu I.
TYP III: VLASTNOSTI
Osteogenesis imperfecta typu III má následující vlastnosti:
- Ačkoli je velmi závažný, nezpůsobuje často smrt v novorozeneckém období;
- Je spojena s „vysokou křehkostí kostí;
- Je zodpovědný za nízký vzrůst, problémy s klouby, svalovou slabost (zejména v nohách a pažích), hrudník, trojúhelníkový obličej a abnormální zakřivení páteře;
- Je to kvůli modré, fialové nebo šedé skléře;
- Může způsobit problémy s dýcháním, křehkost zubů a ztrátu sluchu;
- Často je zodpovědný za deformace kostí;
- Je spojena s kvalitativními a kvantitativními abnormalitami kolagenu typu I.
TYP IV: VLASTNOSTI
Osteogeneze typu IV je charakterizována:
- Stupeň křehkosti kostí mezi formami II a III a formou I;
- Kratší než průměrná postava;
- Modré, purpurové nebo šedě zbarvené skléry;
- Deformace kostí mírné / střední entity, mírné abnormality páteře a sudového hrudníku;
- Trojúhelníkový obličej;
- Možná přítomnost křehkosti zubů a ztráty sluchu;
- Přítomnost abnormalit kolagenu typu I.
TYP V: VLASTNOSTI
Osteogenesis imperfecta typu V se v některých ohledech podobá osteogenesis imperfecta typu IV. Má však některé zvláštnosti, kterými jsou:
- Normálně zbarvené skléry;
- Absence křehkosti zubů;
- Tvorba abnormálních kostních mozolů během procesu hojení zlomených kostí;
- Kalcifikace mezikostní membrány, která se nachází mezi poloměrem a ulnou. To zhoršuje pohyblivost předloktí.
TYP VI: VLASTNOSTI
Také osteogenesis imperfecta typu VI je podobná formě IV. K odlišení od posledně jmenovaných existují některé zvláštnosti, včetně vysokých hladin alkalické fosfatázy v krvi a přítomnosti lamel (kostnatých) na některých kostech podobných rybím trnům.
TYP VII: VLASTNOSTI
Symptomaticky se osteogenesis imperfecta typu VII může za určitých okolností podobat typu IV a za jiných okolností typu II.
Mezi zvláštnosti této závažné patologické formy patří:
- Nízkého vzrůstu;
- Přítomnost extrémně krátkého humeru (pažní kosti) a stehenní kosti (stehenní kosti);
- Častá přítomnost deformity kyčle, známá jako coxa vara.
TYP VIII: CHARAKTERISTIKA
Osteogenesis imperfecta typu VIII velmi připomíná formy II a III.
Mezi jeho zvláštní charakteristiky patří následující: závažný růstový deficit, těžká kosterní hypomineralizace a absence (nebo vzácná přítomnost) enzymu prolyl 3-hydroxylázy.
Diagnóza
Obecně diagnostický proces, kterému jsou vystaveni pacienti s podezřelou formou osteogenesis imperfecta, začíná pečlivým fyzickým vyšetřením a pečlivou anamnézou; poté pokračuje „analýzou rodinné anamnézy pacienta a sérií diagnostických zobrazovacích testů (rentgenové záření, CT vyšetření atd.); nakonec končí kvantitativním a kvalitativním hodnocením kolagenu typu I a genetický test.
Dnes existuje možnost diagnostikovat osteogenesis imperfecta i v prenatální fázi vystavením těhotné ženy ultrazvuku.
DŮLEŽITOST CÍLOVÉHO ZKOUŠENÍ A HISTORIE
Lékař, odborník na osteogenesis imperfecta, je velmi často schopen diagnostikovat výše uvedenou nemoc i pouze pomocí fyzického vyšetření a anamnézy. To znamená, že tyto diagnostické testy nemají žádný zanedbatelný význam.
HODNOCENÍ VÝROBY KOLAGENU TYPU I
Kvalitativní a kvantitativní hodnocení kolagenu typu I je zpravidla velmi spolehlivým testem, protože, jak bylo uvedeno, většina případů osteogenesis imperfecta je charakterizována mutacemi v genech, které řídí produkci kolagenu typu 1.
K posouzení množství a kvality kolagenu typu I přítomného na buněčné úrovni u jednotlivce mohou lékaři spoléhat na kožní biopsii nebo konkrétní krevní test.
Oba tyto hodnotící testy jsou poměrně složité a pacient (nebo jeho rodiče) může na výsledky znát několik týdnů.
GENETICKÝ TEST
Díky genetickému testu, který sonduje celou DNA vyšetřovaného jedince, jsou lékaři schopni definitivně stanovit vlastnosti přítomné genetické mutace.
Obecně se provedení genetického testu na veškeré buněčné DNA předpokládá v případě, že hodnocení charakteristik kolagenu typu I neposkytlo požadované výsledky, nebo pokud nejde o mutaci COL1A1 nebo COL1A2, která způsobuje „osteogenesis imperfecta.
PRENATÁLNÍ DIAGNOSTIKA
Prenatální ultrazvuk je velmi užitečný při identifikaci osteogenesis imperfecta typu II a typu III.
Terapie
V současné době neexistuje žádný specifický lék na osteogenesis imperfecta. Jinými slovy, lidé s osteogenesis imperfecta jsou předurčeni žít se zmíněným stavem až do smrti, což je často důsledkem důsledků samotné nemoci.
Nedostatek specifické terapie nevylučuje existenci jiných forem léčby. Ve skutečnosti mezi terapeutické možnosti pacienta s osteogenesis imperfecta patří různé symptomatické terapie; symptomatickými terapiemi rozumíme ošetření schopná zmírnit příznaky, zpomalit průběh nemoci a zabránit (nebo alespoň odložit) nejzávažnější důsledky.
MOŽNÉ SYMPTOMATICKÉ OŠETŘENÍ
V seznamu možných symptomatických ošetření osteogenesis imperfecta vyniká následující:
- Chirurgické vložení nehtů do nejdelších kostí (poznámka: nejnáchylnější ke zlomeninám), které poskytují větší odolnost vůči zlomeninám a deformitám. Tato operace se nazývá rodding intramedulární;
- Konzervativní nebo chirurgická léčba zlomenin a / nebo kostních deformit;
- Zubní péče k ochraně zdraví zubů;
- Terapie tlumící bolest v případě velmi bolestivých mnohočetných zlomenin;
- Fyzioterapie pro prodloužení a posílení svalů Elastický a posilující svalový aparát vám umožňuje předcházet pádům, které by mohly vést k různým zlomeninám kostí;
- Používání pomůcek pro pohyb, včetně invalidních vozíků, rovnátek, berlí atd.
VÝHODY POHYBU
U jedinců s osteogenesis imperfecta lékaři doporučují neustálé cvičení tělesných cvičení a pohybu obecně, protože obě tyto činnosti přispívají k posílení kosterního a svalového systému.
Mezi doporučené sporty patří: plavání, protože je to „fyzická aktivita s malým dopadem na kosterní systém“, a chůze.
VÝHODY ZE ZDRAVÉHO ŽIVOTNÍHO STYLU
Vedení zdravého života, vyhýbání se kouření, pití přebytečného alkoholu, příliš mnoho a špatného jídla atd. Má pro pacienty s osteogenesis imperfecta více než diskrétní zdravotní výhody, protože zpomaluje progresi onemocnění a snižuje křehkost kostí.
SYMPTOMATICKÉ ZPRACOVÁNÍ VE FÁZI EXPERIMENTACE
V současné době lékaři a výzkumní pracovníci hodnotí účinnost některých symptomatických léčeb, včetně léčby růstovým hormonem a intravenózní a orální terapie na bázi bisfosfonátů.
V tuto chvíli výsledky poskytnuté výše zmíněnou vyšetřovací léčbou přinášejí dobré výsledky pro celou lékařskou komunitu.
Prognóza
Osteogenesis imperfecta je nemoc s negativní prognózou, protože je nevyléčitelná, drasticky ohrožuje kvalitu života a v některých případech způsobuje předčasnou smrt postiženého subjektu.
Je však třeba poznamenat, že i díky moderní symptomatické léčbě je mnoho jedinců s mírnou formou osteogenesis imperfecta schopno vést příjemný a spokojený život.
Prevence
V současné době bohužel neexistuje preventivní opatření proti osteogenesis imperfecta.


















.jpg)