Pozorovatelný u každého 68 000–88 000 novorozenců je Apertův syndrom způsoben specifickou mutací genu FGFR2, který má za úkol regulovat fúzi lebečních stehů a vývoj prstů na rukou a nohou.
Pro diagnostiku Apertova syndromu je zásadní fyzikální vyšetření, anamnéza, radiologické vyšetření lebky a prstů na rukou a nohou a nakonec genetický test.
V současné době mohou lidé trpící Apertovým syndromem počítat pouze se symptomatickou léčbou, to znamená, že zmírňují příznaky a vyhýbají se nejzávažnějším komplikacím.
Stručný přehled kraniálních stehů a jejich fúze
Kraniální stehy jsou vazivové klouby, které slouží ke sloučení kostí lebeční klenby (tj. Čelní, časové, temenní a týlní kosti).
Za normálních podmínek probíhá proces fúze kraniálních stehů v postnatálním období, u některých společných prvků začíná ve věku 1–2 roky a u jiných končí ve věku 20 let. Tento dlouhý a kadenční proces fúze umožňuje mozku adekvátně růst a vyvíjet se.
Apertův syndrom vděčí za svou proslulost nejen svému spojení s kraniostenózou, ale také skutečnosti, že souvisí s určitým stupněm syndaktylie, tedy vrozenou anomálií charakterizovanou fúzí jednoho nebo více prstů nebo prstů. chodidla.
Možnost způsobit kraniostenózu a syndaktylii současně činí z Apertova syndromu příklad akrocefalosyndaktylie; v medicíně je „akrocefalosyndaktylie genetický stav, který kombinuje specifické malformace lebky („ acrocephalus “znamená„ od hlavy ke špičce “) s fúzí jednoho nebo více prstů nebo prstů.
Jaké jsou důsledky rané fúze kraniálního stehu?
Pokud, jako v případě Apertova syndromu a dalších souvisejících onemocnění, dojde k fúzi lebečních stehů během prenatálního, perinatálního (*) nebo velmi raného dětství, mozkové orgány, jako je mozek, mozeček a mozkový kmen a smysl, jak oči procházejí změny v růstu i tvaru.
* Poznámka: „perinatální život“ označuje období mezi 27. týdnem těhotenství a prvních 28 dnů po porodu.
Epidemiologie: Jak častý je Apertův syndrom?
Podle statistik se každý z 65 000–88 000 jedinců narodí s Apertovým syndromem.
Věděli jste, že ...
Genetických chorob, které stejně jako Apertův syndrom způsobují kraniosynostózu, je asi 150.
Mezi nimi kromě Apertova syndromu vyniká na důležitosti Crouzonův syndrom, Pfeifferův syndrom a Saethre-Chotzenův syndrom.
Zvědavost
Získaná mutace, která způsobuje Apertův syndrom, je příkladem „mutace de novo“, tedy„ nové mutace zcela postrádající dědičnou povahu “.
Co způsobuje genovou mutaci spojenou s Apertovým syndromem?
Předpoklad: geny přítomné na lidských chromozomech jsou sekvence DNA, které mají za úkol produkovat základní proteiny v biologických procesech nezbytných pro život, včetně buněčného růstu a replikace.
Pokud je prostý mutací (tedy u zdravého člověka), gen FGFR2 zapojený do Apertova syndromu produkuje ve správném množství receptorový protein, nazývaný receptor Fibroblast Growth Factor Receptor 2, který je nezbytný pro označení načasování fúze lebeční stehy a sledovat oddělení prstů na rukou a nohou (jinými slovy, signalizuje, kdy je vhodný čas na fúzi lebečních stehů a reguluje tvorbu prstů na rukou a nohou).
Na druhou stranu, když prochází mutací pozorovanou za přítomnosti Apertova syndromu, gen FGFR2 je hyperaktivní a produkuje výše uvedený receptorový protein v tak obrovských množstvích, že načasování související s fúzí lebečních stehů je změněno (tj. rychleji) a procesy oddělení prstů na rukou a nohou neprobíhají správně.
Kdo je nejvíce ohrožen?
Pokud jde o získané případy Apertova syndromu, faktory, které indukují mutaci genu FGFR2 po početí, nejsou v tuto chvíli absolutně jasné.
Výzkum tohoto aspektu stále pokračuje.
Apertův syndrom je autozomálně dominantní onemocnění
Rozumět...
Každý lidský gen je přítomen ve dvou kopiích, nazývaných alely, jedna mateřského původu a jedna otcovského původu.
Apertův syndrom má všechny vlastnosti autozomálně dominantní choroby.
Genetická nemoc je autozomálně dominantní, když se mutace jediné kopie genu, která ji způsobuje, stačí projevit.
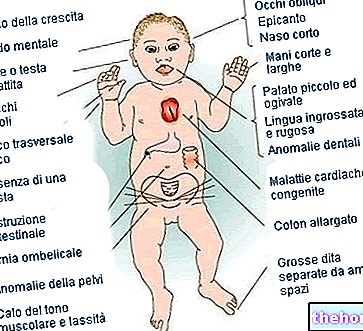
- Plochý nebo konkávní obličej (v důsledku nedostatečného růstu centrálních kostí obličeje)
- Nafouklé, vypoulené a široce otevřené oči mělké oční důlky a abnormálně rozmístěné oči (hypertelorismus očních důlků);
- Zobákový nos;
- Nedostatečně vyvinutá čelist v kombinaci s prognatismem;
- Přeplněné zuby (kvůli nedostatečně vyvinuté čelisti)
- Uši nižší než normálně.
Syndaktylie
U nosičů Apertova syndromu je syndaktylie pozorována v rukou, téměř vždy a na nohou, méně často než v rukou.

Typické charakteristiky syndaktylie v rukou jedince s Apertovým syndromem jsou 4:
- Přítomnost krátkého palce s radiální odchylkou (tj. Orientovaná abnormálně směrem k poloměru, jedné ze dvou kostí předloktí);
- Složitá syndaktylie mezi ukazováčkem, prostředníkem a prsteníčkem. Komplexní syndaktylií lékaři myslí abnormální spojení prstů, které postihuje nejen měkké tkáně (kůži), ale také kostní tkáně (falangy);
- Symfalangismus. Je to lékařský termín, který označuje anomální fúzi interfalangeálních kloubů prstů (interfalangeální klouby jsou kloubní prvky přítomné mezi falangou a falangou);
- Jednoduchá syndaktylie mezi čtvrtým a pátým prstem (tj. Mezi prstenem a malíčkem). U jednoduché syndaktylie odborníci označují abnormální spojení prstů, které postihuje pouze měkké tkáně (kůži).
SEVERITY SYNDROME IN OPEN SYNDROME: 3 TYPY
Na základě závažnosti malformace palce (první ze čtyř charakteristik) odborníci na Apertův syndrom rozlišují 3 typy syndaktylie rostoucí závažnosti:
- Typ I (nejméně závažný) se shoduje s „minimální anomálií postihující palec, která zůstává zcela nezávislá na indexu“.
Jiné anomálie: ukazováček, prostředník a prsteníček jsou spojeny dohromady složitou syndaktylií a přítomným symfalangismem postihujícím distální interfalangeální klouby; c “je jednoduchá a neúplná syndaktylie mezi prstenem a malíčky (neúplná syndaktylie znamená, že fúze mezi dvěma prsty je částečná).
Jiná informace: je nejběžnější typ. - Typ II (střední závažnost) je charakterizován výraznější radiální odchylkou palce ve srovnání s předchozím případem a zásadou syndaktylie mezi palcem a ukazováčkem (c "je neúplná syndaktylie mezi palcem a ukazováčkem) .
Jiné anomálie: indexové, prostřední a prstencové prsty jsou protagonisty komplexní syndaktylie kombinované s distálním symfalangismem; mezi prsteníčkem a malíčkem c “je jednoduchá a neúplná syndaktylie.
Jiná informace: je to druhý nejběžnější typ. - Typ III (nejtěžší) je charakterizován přítomností palce plně spojeného s indexem, a to nejen na úrovni měkkých tkání, ale také na úrovni kostních tkání.
Jiné anomálie: všechny prsty jsou spojeny dohromady, a to natolik, že je téměř nemožné je rozpoznat; c "je" jeden hřebík; pokud je mezi prvními 4 prsty syndaktylie složitá, mezi prsteníkem a malíčkem je (jako u ostatních typů) jednoduchá a neúplná.
Jiná informace: je to nejvzácnější typ.
Další možné příznaky a příznaky Apertova syndromu
V některých případech, kromě toho, že je Apertův syndrom spojován s kraniosynostózou a syndaktylií, souvisí s přítomností: polydaktylie (tj. Přítomnosti prstu navíc v rukou nebo nohou), ztráty sluchu, recidivujícího ucha a dutin, hyperhidrózy, mastnoty kůže, silné akné, žádné vlasy na obočí, fúze krčních obratlů, syndrom obstrukční spánkové apnoe a / nebo rozštěp patra.
Komplikace
Komplikace Apertova syndromu jsou především vážnými důsledky, které může mít kraniosynostóza na vývoj mozku a intelektuálních schopností a na funkční schopnosti rukou podléhajících syndaktylii.
Kdy je možné detekovat Apertův syndrom?
Kraniální a digitální abnormality způsobené Apertovým syndromem jsou obvykle patrné při narození, takže diagnostika a plánování léčby jsou okamžité.
do hlavy (rentgenové snímky hlavy, CT hlavy a / nebo MRI hlavy) a rukou a případně nohou; nakonec to končí genetickým testem.
Fyzikální vyšetření a anamnéza
Fyzikální vyšetření a anamnéza v zásadě spočívají v přesném vyšetření symptomů vykazovaných pacientem.
V kontextu Apertova syndromu lékař v těchto okamžicích diagnostického procesu zjišťuje kraniosynostózu a syndaktylii a jejich přesné charakteristiky.
Radiologická vyšetření hlavy a prstů na rukou a nohou
V kontextu Apertova syndromu:
- Radiologická vyšetření hlavy lékař používá k potvrzení přítomnosti časné fúze koronárních stehů (koronální kraniosynostóza nebo brachycefalie); navíc mu umožňují odhadnout závažnost současných kranio-encefalických anomálií.
- Na druhé straně jsou radiologická vyšetření prstů na rukou a nohou nezbytná ani ne tak pro potvrzení syndaktylie (k tomu stačí vizuální vyšetření), jako spíše pro detailní poznání charakteristik interdigitálních fúzí (typ syndaktylie, úroveň) fúze atd.).
Genetický test
Jedná se o analýzu DNA zaměřenou na detekci mutací v kritických genech.
V kontextu Apertova syndromu představuje potvrzující diagnostický test, protože přináší na světlo charakteristiku mutace FGFR2 pro dané genetické onemocnění.
OPERAČNÍ PÉČE O BRACHYCEFALII
U nosiče Apertova syndromu zahrnuje chirurgická léčba brachycefalie:
- První intervence v mladém věku (do roku života), zaměřená na oddělení koronálních prošívacích stehů dříve, než se očekávalo. Pokud je tato intervence úspěšná, mozek si užívá správný prostor pro růst a snižuje se riziko intelektuálních problémů.
- Druhá intervence ve věku od 4 do 12 let zaměřená na normální vzhled obličeje, která (jak si čtenář vzpomene) je plochá, ne -li konkávní.
Dotyčná operace zahrnuje řez některých kostí obličeje a jejich přemístění podle uspořádání, které alespoň částečně odráží normálnost. - Třetí případný zásah v dětství s cílem eliminovat nebo alespoň omezit oční hypertelorismus.
CHIRURGICKÁ PÉČE O SYNDACI
Chirurgická léčba syndaktylie se liší podle charakteristik interdigitální fúze (závisí tedy na typu).

To znamená, že intervence platná pro jedince s Apertovým syndromem nemusí být stejně platná pro jiného jedince se stejným genetickým onemocněním (platí pouze tehdy, je -li typ přítomné syndaktylie stejný).
Po vyjasnění tohoto základního aspektu je cíl každého typu stávajícího chirurgického přístupu stejný a spočívá v uvolnění srostlých prstů, aby byla zaručena určitá funkčnost rukou.
Léčba syndaktylie obecně zahrnuje dvě fáze:
- 1 krok: „uvolněte“ první interdigitální prostor (prostor mezi palcem a ukazováčkem) a čtvrtý interdigitální prostor (prostor mezi prsteníčkem a malíčkem);
- 2 kroky: „uvolněte“ druhý a třetí interdigitální prostor (prostor mezi ukazováčkem a prostředníkem a prostor mezi prostředníkem a prsteníkem).


















.jpg)