Aktivní složky: Temozolomid
Temodal 5 mg tvrdé tobolky
Temodal 20 mg tvrdé tobolky
Temodal 100 mg tvrdé tobolky
Temodal 140 mg tvrdé tobolky
Temodal 180 mg tvrdé tobolky
Temodal 250 mg tvrdé tobolky
Proč se přípravek Temodal používá? K čemu to je?
Temodal obsahuje léčivý přípravek zvaný temozolomid. Tento lék je protirakovinným činidlem.
Temodal se používá k léčbě specifických forem mozkových nádorů:
- u dospělých s prvním diagnostikovaným multiformním glioblastomem. Temodal se zpočátku používá v kombinaci s radioterapií (fáze souběžné léčby) a následně samostatně (fáze léčby monoterapií).
- u dětí ve věku 3 let a starších a u dospělých pacientů s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom. Temodal se používá u těchto nádorů, které po standardní terapii recidivují nebo progredují.
Kontraindikace Kdy by Temodal neměl být používán
Neužívejte Temodal
- jestliže jste alergický (á) na temozolomid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže jste v minulosti měl (a) alergickou reakci na dakarbazin (protinádorový lék, někdy nazývaný DTIC). Mezi příznaky alergické reakce patří svědění, dušnost nebo sípání, otok obličeje, rtů, jazyka nebo hrdla.
- jestliže je počet určitých typů krvinek výrazně snížen (myelosuprese), jako je počet bílých krvinek a krevních destiček. Tyto krvinky jsou důležité pro boj s infekcemi a pro správnou srážlivost krve. Váš lékař vám provede krevní testy, aby se ujistil, že je dostatek buněk pro zahájení léčby.
Opatření pro použití Co potřebujete vědět před užitím přípravku Temodal
Před užitím přípravku Temodal se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou,
- musí být pečlivě sledován pro rozvoj závažné formy infekce hrudníku zvané Pneumocystis jirovecii pneumonia (PCP). Pokud jste poprvé diagnostikovaným pacientem (multiformní glioblastom), můžete dostat Temodal po dobu 42 dnů v kombinaci s radioterapií. V takovém případě vám lékař také předepíše léky, které vám pomohou předcházet tomuto typu zápalu plic (PCP).
- pokud jste někdy měli nebo v současné době můžete mít infekci hepatitidou B. Důvodem je, že Temodal může způsobit reaktivaci hepatitidy B, což v některých případech může být smrtelné. Před zahájením léčby bude pacient pečlivě zkontrolován lékařem, aby zkontroloval, zda existují známky této infekce.
- jestliže máte nízký počet červených krvinek (anémie), bílých krvinek a krevních destiček nebo máte problémy se srážlivostí krve před zahájením léčby nebo se u nich objeví během léčby. Váš lékař se může rozhodnout snížit dávku, ukončit, zastavit nebo změnit Můžete také potřebovat další léčbu. V některých případech možná budete muset přestat užívat Temodal. Vzorky vaší krve budou během léčby často testovány, aby se zkontrolovaly nežádoucí účinky přípravku Temodal na vaše krvinky.
- protože můžete mít nízké riziko vzniku dalších poruch krevních buněk, včetně leukémie.
- pokud máte nevolnost (pocit nevolnosti v žaludku) a / nebo zvracení, což jsou velmi časté nežádoucí účinky přípravku Temodal (viz bod 4), může vám lékař předepsat lék (antiemetikum), který pomůže předcházet zvracení. Pokud často zvracíte před léčbou nebo během léčby, požádejte svého lékaře o nejvhodnější dobu užívání přípravku Temodal, dokud nebude zvracení pod kontrolou. Pokud po užití jedné dávky zvracíte, druhou už ve stejný den neberte.
- pokud máte horečku nebo příznaky infekce, okamžitě kontaktujte svého lékaře.
- pokud je vám více než 70 let, můžete být náchylnější k infekcím, tvorbě modřin nebo krvácení.
- pokud máte problémy s játry nebo ledvinami, bude možná nutné upravit dávku přípravku Temodal.
Děti a dospívající
Nepodávejte tento přípravek dětem mladším 3 let, protože nebyl studován. U pacientů starších 3 let, kteří užívali přípravek Temodal, jsou k dispozici pouze omezené informace.
Interakce Které léky nebo potraviny mohou změnit účinek přípravku Temodal
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval (a) nebo které možná budete užívat.
Varování Je důležité vědět, že:
Těhotenství, kojení a plodnost
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat. Důvodem je, že by neměl být léčen přípravkem Temodal v těhotenství, pokud to lékař výslovně neurčí.
Pacienti mužského i ženského pohlaví, kteří užívají Temodal, by měli používat účinná antikoncepční opatření (viz také „Mužská plodnost“ níže).
Pokud jste léčena přípravkem Temodal, musíte přestat kojit.
Mužská plodnost
Temodal může způsobit trvalou neplodnost. Pacienti mužského pohlaví by měli používat účinné metody antikoncepce a nesnažit se mít dítě 6 měsíců po ukončení léčby.Před zahájením léčby se doporučuje informovat se o skladování spermií.
Řízení dopravních prostředků a obsluha strojů
Temodal může způsobit, že se budete cítit unavení nebo ospalí. V takovém případě neřiďte ani nepoužívejte žádné nástroje nebo stroje nebo jízdní kola, dokud neuvidíte účinky, které na vás tento lék má (viz bod 4).
Temodal obsahuje laktózu
Temodal obsahuje laktózu (druh cukru). Pokud vám lékař řekl, že trpíte „nesnášenlivostí některých cukrů, kontaktujte svého lékaře před užitím tohoto léku.
Dávkování a způsob použití Jak používat Temodal: Dávkování
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud máte pochybnosti, poraďte se se svým lékařem nebo lékárníkem.
Dávkování a trvání léčby
Váš lékař určí vaši dávku přípravku Temodal. Záleží na vaší velikosti (výšce a hmotnosti), a pokud máte recidivující nádor, na jakékoli předchozí léčbě chemoterapií.
Mohou být předepsány další léky (antiemetika), které je třeba užívat před a / nebo po užití přípravku Temodal k prevenci nebo kontrole nevolnosti a zvracení.
Pacienti s multiformním glioblastomem nejprve diagnostikovali:
Pokud jste pacient, kterému byla poprvé diagnostikována rakovina, léčba bude probíhat ve dvou fázích:
- Temodal je zpočátku spojen s radioterapií (souběžná fáze)
- Poté se přípravek Temodal podává samostatně (fáze monoterapie).
Během souběžné fáze zahájí lékař Temodal v dávce 75 mg / m2 (obvyklá dávka). Tuto dávku budete užívat každý den po dobu 42 dnů (maximálně 49 dní) v kombinaci s radioterapií. Vaše dávka přípravku Temodal může být zpožděna nebo zastavena na základě počtu vašich krvinek a toho, jak se s přípravkem vyrovnáváte během souběžné fáze.
Jakmile je radioterapie dokončena, léčbu na 4 týdny ukončíte. To dá vašemu tělu šanci na zotavení.
Poté zahájí monoterapii.
Během fáze monoterapie se bude dávka a způsob užívání přípravku Temodal lišit. Váš lékař určí vaši správnou dávku. Může existovat až 6 léčebných období (cyklů). Každý trvá 28 dní. Svou novou dávku přípravku Temodal budete užívat samostatně jednou denně po dobu prvních 5 dnů („terapeutické dny“) každého cyklu. První dávka bude 150 mg / m 2. Poté bude následovat 23 dní bez Temodalu. Tím je dosaženo 28 dnů léčebného cyklu.
Po 28. dnu začne další cyklus. Znovu budete užívat přípravek Temodal jednou denně po dobu 5 dnů a poté 23 dní bez přípravku Temodal. Dávku přípravku Temodal lze upravit, oddálit nebo ukončit na základě krevního obrazu a toho, jak se s léčivým přípravkem vyrovnáváte během každého léčebného cyklu.
Pacienti s nádory, které se vrátily nebo se zhoršily (maligní gliomy, jako je multiformní glioblastom nebo anaplastický astrocytom) léčené samotným přípravkem Temodal:
Kurz léčby přípravkem Temodal trvá 28 dní. Temodal budete užívat samostatně jednou denně po dobu prvních 5 dnů. Tato denní dávka závisí na jakékoli předchozí léčbě chemoterapií.
Pokud jste dříve nebyli léčeni chemoterapií, bude vaše první dávka přípravku Temodal 200 mg / m2 jednou denně po dobu prvních 5 dnů. Pokud jste byli dříve léčeni chemoterapií, bude vaše první dávka přípravku Temodal 150 mg / m 2 jednou denně po dobu prvních 5 dnů.
Dalších 23 dní již Temodal nedostanete. Tím bude dokončen 28denní cyklus.
Po 28. dnu začne další cyklus. Znovu budete užívat Temodal jednou denně po dobu pěti dnů, poté následuje 23 dní bez Temodalu.
Před každým novým cyklem vám budou provedeny krevní testy, aby se zjistilo, zda je třeba dávku přípravku Temodal změnit. Na základě výsledků vašich krevních testů vám lékař může změnit dávku pro další cyklus.
Jak se přípravek Temodal užívá
Užívejte předepsanou dávku přípravku Temodal jednou denně, nejlépe každý den ve stejnou dobu.
Vezměte kapsle na prázdný žaludek; například alespoň jednu hodinu před snídaní.
Polkněte tobolku (y) celou se sklenicí vody. Neotvírejte, nedrťte ani nežvýkejte tobolky. Pokud je tobolka poškozená, vyvarujte se kontaktu prášku s kůží, očima nebo nosem. Nějaký prášek by měl přijít do styku s vaším oči nebo nos, opláchněte postižené místo vodou.
V závislosti na předepsané dávce může být nutné užít více než jednu tobolku současně, případně s různou silou (obsah účinné látky v mg). Barva pláště tobolky je pro každou sílu jiná (viz tabulka níže).
Musíte se ujistit, že rozumíte a pamatujete si následující:
- kolik kapslí potřebujete k denní dávce. Požádejte svého lékaře nebo lékárníka, aby to zapsal (včetně barvy).
- jaké jsou dny užívání terapie.
Zkontrolujte dávku u svého lékaře pokaždé, když budete muset zahájit nový cyklus, protože se může lišit od předchozího cyklu.
Vždy užívejte Temodal přesně podle pokynů svého lékaře. Pokud máte pochybnosti, poraďte se se svým lékařem nebo lékárníkem. Chyby při užívání tohoto léku by mohly způsobit vážné poškození zdraví.
Jestliže jste zapomněl (a) užít Temodal
Užijte vynechanou dávku co nejdříve ve stejný den. Pokud uplynul celý den, kontaktujte svého lékaře. Nezdvojnásobujte následující dávku, abyste nahradil (a) zapomenutou dávku, pokud vám to lékař neřekne.
Máte -li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Předávkování Co dělat, když jste užil příliš mnoho přípravku Temodal
Pokud omylem užijete více tobolek, než je předepsáno, kontaktujte ihned svého lékaře, lékárníka nebo zdravotní sestru.
Nežádoucí účinky Jaké jsou vedlejší účinky přípravku Temodal
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Okamžitě kontaktujte svého lékaře, pokud zaznamenáte:
- závažná alergická (hypersenzitivní) reakce (kopřivka, sípání nebo jiné potíže s dýcháním),
- nekontrolované krvácení,
- záchvaty (křeče),
- horečka,
- silná bolest hlavy, která nezmizí.
Léčba přípravkem Temodal může způsobit snížení počtu určitých typů krvinek. To může vést ke zvýšené tvorbě modřin nebo krvácení, anémii (snížení počtu červených krvinek), horečce a snížení odolnosti vůči infekcím. Snížení počtu krvinek je obvykle krátkodobé. V některých případech může být prodloužena a může vést k velmi těžké formě anémie (aplastické anémie). Váš lékař vám bude pravidelně kontrolovat krev a rozhodne, zda je nutná specifická terapie. V některých případech bude dávka přípravku Temodal snížena nebo léčba ukončena.
Nežádoucí účinky z klinických studií:
Temodal v kombinované léčbě s radioterapií u nově diagnostikovaného glioblastomu
U pacientů užívajících přípravek Temodal v kombinaci s radioterapií se mohou objevit jiné nežádoucí účinky, než jaké byly hlášeny u pacientů, kteří dostávali přípravek Temodal samotný. Mohou se objevit následující nežádoucí účinky, které mohou vyžadovat lékařskou pomoc.
Velmi časté (mohou postihnout více než 1 z 10 lidí):
ztráta chuti k jídlu, bolest hlavy, zácpa (potíže s vyprazdňováním stolice), nevolnost (pocit nevolnosti v žaludku), zvracení, vyrážka, ztráta vlasů, únava.
Časté (mohou postihnout až 1 z 10 lidí):
infekce úst, infekce ran, snížený počet krvinek (neutropenie, trombocytopenie, lymfopenie, leukopenie), zvýšení hladiny cukru v krvi, úbytek na váze, změny duševního stavu nebo bdělosti, úzkost / deprese, ospalost, potíže s mluvením, problémy s rovnováhou, závratě, zmatenost ztráta paměti, potíže se soustředěním, neschopnost usnout nebo usnout, pocit brnění, podlitiny, třes, abnormální nebo zmatené vidění, dvojité vidění, zhoršený sluch, dušnost, kašel, krevní sraženiny v nohou, zadržování tekutin, otoky nohou průjem, bolest žaludku nebo břicha, pálení žáhy, žaludeční nevolnost, potíže s polykáním, sucho v ústech, podráždění nebo zarudnutí kůže, suchá kůže, svědění, ochablost svalů, bolest kloubů, bolesti svalů a bolesti, časté močení, potíže s držením moči, alergická reakce, horečka, poranění radiového kola pia, otok obličeje, bolest, změna chuti, abnormální testy jaterních funkcí.
Méně časté (mohou postihnout až 1 ze 100 lidí):
příznaky chřipky, podkožní červené skvrny, nízká hladina draslíku v krvi, zvýšení tělesné hmotnosti, změny nálady, halucinace a poruchy paměti, částečná paralýza, poruchy koordinace, smyslové poruchy, částečná ztráta zraku, suché nebo bolestivé oči, hluchota, infekce středního ucha, zvonění uši, bolest ucha, bušení srdce (když slyšíte tlukot srdce), krevní sraženiny v plicích, vysoký krevní tlak, zápal plic, zánět nosních cest, bronchitida, nachlazení nebo chřipka, nadýmání žaludku, potíže s ovládáním pohybu střev, hemoroidy, olupování kůže zvýšená citlivost kůže na sluneční světlo, změny barvy kůže, zvýšené pocení, poškození svalů, bolest zad, potíže s močením, vaginální krvácení, sexuální impotence, chybějící nebo silné menstruace, vaginální podráždění, bolest prsou, návaly horka, zimnice i, změna barvy jazyka, změna vnímání pachů, žízeň, zubní poruchy.
Temodal sám o sobě v gliomu, který se vrátil nebo se zhoršil
Mohou se objevit následující nežádoucí účinky, které mohou vyžadovat lékařskou pomoc.
Velmi časté (mohou postihnout více než 1 z 10 lidí):
snížený počet krvinek (neutropenie nebo lymfopenie, trombocytopenie), ztráta chuti k jídlu, bolest hlavy, zvracení, nevolnost (pocit nevolnosti v žaludku), zácpa (potíže s stolicí), únava.
Časté (mohou postihnout až 1 z 10 lidí):
ztráta hmotnosti, ospalost, závratě, pocit brnění, dušnost, průjem, bolest břicha, žaludeční nevolnost, vyrážka, svědění, ztráta vlasů, horečka, slabost, zimnice, pocit nevolnosti, bolest, změna chuti.
Méně časté (mohou postihnout až 1 ze 100 lidí):
snížení počtu krvinek (pancytopenie, anémie, leukopenie).
Vzácné (mohou postihnout až 1 z 1000 lidí):
kašel, infekce včetně zápalu plic.
Velmi vzácné (mohou postihnout až 1 z 10 000 lidí):
zarudnutí kůže, kopřivka, vyrážka, alergické reakce.
Další nežádoucí účinky:
Často byly hlášeny případy zvýšení jaterních enzymů. Méně často byly hlášeny případy zvýšeného bilirubinu, problémy s odtokem žluči (cholestáza), hepatitida a poškození jater, včetně selhání jater s následkem smrti.
Byly pozorovány velmi vzácné případy závažné vyrážky s otokem kůže, včetně na dlaních a chodidlech nebo bolestivého zarudnutí kůže a / nebo puchýřů na těle nebo v ústech. Pokud se tyto případy vyskytnou, okamžitě to sdělte svému lékaři.
U přípravku Temodal byly pozorovány velmi vzácné případy plicních nežádoucích účinků. Pacienti se obvykle dostavují s dušností a kašlem. Informujte svého lékaře, pokud si všimnete některého z těchto příznaků.
Ve velmi vzácných případech mohou mít pacienti užívající přípravek Temodal a podobné léky malé riziko vzniku sekundárních nádorových onemocnění, včetně leukémie.
Méně často byly hlášeny nové nebo reaktivované (rekurentní) cytomegalovirové infekce a reaktivované infekce virem hepatitidy B.
Méně často byly hlášeny případy diabetes insipidus. Mezi příznaky diabetes insipidus patří vylučování velkého množství moči a pocit žízně.
Hlášení nežádoucích účinků
Pokud se u vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. To se týká i všech možných nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. poskytnout více informací o bezpečnosti tohoto léku.
Expirace a retence
Uchovávejte tento přípravek mimo dohled a dosah dětí, nejlépe v uzamčené skříňce. Náhodné požití může způsobit smrt dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku a krabičce. Datum exspirace se vztahuje k poslednímu dni uvedeného měsíce.
Prezentace lahví
Uchovávejte při teplotě do 30 ° C.
Tobolky uchovávejte v původní lahvičce, aby byly chráněny před vlhkostí.
Láhev uchovávejte těsně uzavřenou.
Prezentace ve sáčku
Uchovávejte při teplotě do 30 ° C.
Informujte svého lékárníka, pokud si všimnete jakýchkoli změn ve vzhledu tobolek.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Pomůže to chránit životní prostředí.
Jiná informace
Co Temodal obsahuje
- Léčivou látkou je temozolomid.
- Temodal 5 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 5 mg.
- Temodal 20 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 20 mg.
- Temodal 100 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 100 mg.
- Temodal 140 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 140 mg.
- Temodal 180 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 180 mg.
- Temodal 250 mg tvrdé tobolky: Jedna tobolka obsahuje temozolomidum 250 mg.
Dalšími složkami jsou:
- obsah kapslí: bezvodá laktóza, koloidní bezvodý oxid křemičitý, sodná sůl karboxymetylškrobu typu A, kyselina vinná, kyselina stearová (viz bod 2 „Temodal obsahuje laktózu“).
- obal tobolky:
- Temodal 5 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), laurylsulfát sodný, žlutý oxid železitý (E 172), indigokarmín (E 132),
- Temodal 20 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), laurylsulfát sodný, žlutý oxid železitý (E 172),
- Temodal 100 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), laurylsulfát sodný, červený oxid železitý (E 172),
- Temodal 140 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), laurylsulfát sodný, indigokarmín (E 132),
- Temodal 180 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), laurylsulfát sodný, žlutý oxid železitý (E 172) a červený oxid železitý (E 172),
- Temodal 250 mg tvrdé tobolky: želatina, oxid titaničitý (E 171), laurylsulfát sodný. tiskařský inkoust: šelak, propylenglykol, čištěná voda, hydroxid amonný, hydroxid draselný a černý oxid železitý (E 172).
Popis toho, jak Temodal vypadá a obsah balení
- Temodal 5 mg tvrdé tobolky mají neprůhledné bílé tělo, neprůhledné zelené víčko a jsou potištěny černým inkoustem.
- Temodal 20 mg tvrdé tobolky mají neprůhledné bílé tělo, neprůhledné žluté víčko a jsou potištěny černým inkoustem.
- Temodal 100 mg tvrdé tobolky mají neprůhledné bílé tělo, neprůhledné růžové víčko a jsou potištěny černým inkoustem.
- Temodal 140 mg tvrdé tobolky mají neprůhledné bílé tělo, modré víčko a jsou potištěny černým inkoustem.
- Temodal 180 mg tvrdé tobolky mají neprůhledné bílé tělo, neprůhledné oranžové víčko a jsou potištěny černým inkoustem.
- Temodal 250 mg tvrdé tobolky mají neprůhledné bílé tělo a víčko a jsou potištěny černým inkoustem.
Prezentace lahví
Tvrdé tobolky pro perorální podání jsou k dispozici v lahvích ze žlutého skla obsahujících 5 nebo 20 tobolek.
Krabice obsahuje jednu láhev.
Prezentace ve sáčku
Tvrdé tobolky pro perorální podání jsou k dispozici v krabičkách obsahujících 5 nebo 20 tvrdých tobolek, které jsou jednotlivě uzavřeny ve sáčcích.
Na trhu nemusí být všechny velikosti balení.
Zdroj příbalové informace: AIFA (Italská agentura pro léčivé přípravky). Obsah zveřejněný v lednu 2016. Přítomné informace nemusí být aktuální.
Abyste měli přístup k nejaktuálnější verzi, doporučujeme navštívit webovou stránku AIFA (Italská agentura pro léčivé přípravky). Prohlášení a užitečné informace.
01.0 NÁZEV LÉČIVÉHO PŘÍPRAVKU
TEMODAL 100 MG TVRDÉ Kapsle
02.0 KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje temozolomidum 100 mg.
Pomocná látka se známými účinky:
Jedna tvrdá tobolka obsahuje 175,7 mg bezvodé laktózy.
Úplný seznam pomocných látek viz bod 6.1.
03.0 LÉKOVÁ FORMA
Tvrdá tobolka (kapsle).
Tvrdé tobolky mají neprůhledné bílé tělo, neprůhledné růžové víčko a jsou potištěny černým inkoustem.
Na obalu je vytištěno „Temodal“. Na těle je vytištěno „100 mg“, logo Schering-Plough a dva pruhy.
04.0 KLINICKÉ INFORMACE
04.1 Terapeutické indikace
Temodal je indikován k léčbě:
• dospělí pacienti s nejprve diagnostikovaným multiformním glioblastomem v kombinaci s radioterapií (RT) a později jako monoterapie.
• dětští pacienti ve věku ≥ 3 roky, mladiství a dospělí s maligním gliomem, jako je multiformní glioblastom nebo anaplastický astrocytom, u nichž po standardní terapii dojde k relapsu nebo progresi.
04.2 Dávkování a způsob podání
Temodal by měli předepisovat pouze lékaři se zkušenostmi s onkologickou léčbou mozkových nádorů.
Může být podána antiemetická léčba (viz bod 4.4).
Dávkování
Dospělí pacienti s prvním diagnostikovaným multiformním glioblastomem
Temodal se podává v kombinaci s fokální radioterapií (souběžná fáze) a následně jako monoterapie až 6 cyklů temozolomidu (TMZ) (fáze monoterapie).
Souběžná fáze
TMZ se podává orálně v denní dávce 75 mg / m2 po dobu 42 dnů v kombinaci s fokální radioterapií (60 Gy podávaných ve 30 frakcích). Nedoporučuje se žádné snižování dávky, ale na základě kritérií hematologické a nehematologické toxicity bude týdenní rozhodování, zda podávání TMZ odložit nebo přerušit. V podávání TMZ lze pokračovat během 42denního souběžného období (maximálně 49 dní), pokud jsou splněny všechny následující podmínky:
• absolutní počet neutrofilů (ANC) ≥ 1,5 x 109 / l
• počet trombocytů ≥ 100 x 109 / l
• Společná kritéria toxicity (CTC) pro nehematologickou toxicitu ≤ stupeň 1 (kromě alopecie, nevolnosti a zvracení).
Během léčby by měl být každý týden prováděn kompletní krevní obraz. Léčba TMZ by měla být dočasně nebo trvale přerušena během souběžné fáze na základě kritérií hematologické a nehematologické toxicity, jak je uvedeno v tabulce 1.
a: Souběžná léčba TMZ může pokračovat, pokud jsou splněny všechny následující podmínky: absolutní počet neutrofilů ≥ 1,5 x 109 / l; počet trombocytů ≥ 100 x 109 / l; CTC nehematologická toxicita ≤ stupeň 1 (kromě alopecie, nauzey, zvracení).
Fáze monoterapie
Čtyři týdny po ukončení souběžné fáze TMZ + RT se TMZ podává až 6 cyklů jako monoterapie. Dávka cyklu 1 (monoterapie) je 150 mg / m2 jednou denně po dobu 5 dnů, po nichž následuje 23 dní bez léčby. Na začátku cyklu 2 se dávka zvýší na 200 mg / m2, pokud je CTC pro nehematologickou toxicitu pro cyklus 1 stupeň ≤ 2 (kromě alopecie, nauzey a zvracení), absolutní počet neutrofilů (ANC) je ≥ 1,5 x 109 / l a počet trombocytů je ≥ 100 x 109 / l. Pokud není dávka zvýšena na cyklus 2, nelze v následujících cyklech dávku zvýšit. Jakmile je dávka zvýšena, dávka zůstane na 200 mg / m2 na den po dobu prvních 5 dnů každého následujícího cyklu, pokud nedojde k toxicitě. Snížení dávky a přerušení léčby během fáze monoterapie by mělo být provedeno v souladu s tabulkami 2 a 3.
Během léčby by měl být 22. den (21 dní po první dávce TMZ) proveden kompletní krevní obraz. Dávka by měla být snížena nebo podávání přerušeno podle tabulky 3.
Tabulka 2. Úrovně dávek monoterapie TMZ
Tabulka 3. Snížení dávky TMZ nebo přerušení během monoterapie
a: Úrovně dávek TMZ jsou uvedeny v tabulce 2.
b: TMZ musí být přerušeno, pokud:
• úroveň dávky -1 (100 mg / m2) stále způsobuje nepřijatelnou toxicitu
• ke stejné nehematologické toxicitě 3. stupně stále dochází po snížení dávky (kromě alopecie, nauzey, zvracení).
Dospělí a dětští pacienti ve věku nejméně 3 roky s rekurentním nebo progresivním maligním gliomem:
Terapie zahrnuje 28denní léčebný cyklus. U pacientů, kteří dříve nepodstoupili chemoterapii, se TMZ podává orálně v dávce 200 mg / m2 jednou denně po dobu prvních 5 dnů s následným „přerušením léčby na 23 dní (celkový 28denní léčebný cyklus)“. podstupující chemoterapii je počáteční dávka 150 mg / m2 jednou denně, která se ve druhém cyklu zvýší na 200 mg / m2 jednou denně po dobu 5 dnů bez hematologické toxicity (viz bod 4.4).
Zvláštní populace
Pediatrická populace
U pacientů ve věku 3 let nebo starších by měl být TMZ používán pouze u rekurentního nebo progresivního maligního gliomu. Zkušenosti s těmito dětmi jsou velmi omezené (viz body 4.4 a 5.1) Bezpečnost a účinnost TMZ u dětí mladších 3 let nebyla stanovena. Nejsou k dispozici žádné údaje.
Pacienti s poruchou funkce jater nebo ledvin
Farmakokinetika TMZ u pacientů s normální funkcí jater je srovnatelná s farmakokinetikou pacientů se středně těžkou nebo středně těžkou poruchou funkce jater. Nejsou k dispozici žádné údaje o podávání TMZ u pacientů s těžkou poruchou funkce jater (dětská třída C) nebo poruchou funkce ledvin. Na základě farmakokinetických vlastností TMZ je nepravděpodobné, že by bylo nutné snížení dávky u pacientů s těžkou poruchou funkce jater nebo s jakýmkoli stupněm poruchy funkce ledvin. TMZ by však měl být u těchto pacientů podáván s opatrností.
Starší pacienti
Farmakokinetická analýza na populaci pacientů ve věku 19 až 78 let ukázala, že clearance TMZ není ovlivněna věkem. U starších pacientů (ve věku> 70 let) se však zdá, že existuje zvýšené riziko neutropenie a trombocytopenie (viz bod 4.4).
Způsob podání
Temodal tvrdé tobolky se užívají na prázdný žaludek.
Tobolky se polykají celé a zapíjejí se sklenicí vody a nesmí se otevírat ani žvýkat.
Pokud po podání dávky dojde ke zvracení, nelze stejnou dávku podat ve stejný den.
04.3 Kontraindikace
Přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Přecitlivělost na dakarbazin (DTIC).
Těžká myelosuprese (viz bod 4.4).
04.4 Zvláštní upozornění a vhodná opatření pro použití
Oportunistické infekce a reaktivace infekcí
Během léčby TMZ byly pozorovány oportunní infekce (jako je pneumonie Pneumocystis jirovecii) a reaktivace infekcí (jako HBV, CMV) (viz bod 4.8).
Zápal plic z Pneumocystis jirovecii
U pacientů, kteří dostávali TMZ v kombinaci s RT v pilotní studii po 42denním programu prodloužené léčby, bylo prokázáno, že jsou zvláště ohroženi rozvojem zápalu plic Pneumocystis jirovecii (PCP). V důsledku toho je u všech pacientů, kteří dostávají TMZ a RT souběžně po dobu 42 dnů (s maximem 49 dnů), bez ohledu na počet lymfocytů, nutná profylaxe PCP. Pokud dojde k lymfopenii, pacienti by měli pokračovat v profylaxi, dokud lymfopenie neklesne na stupeň ≤ 1.
Širší recidivu PCP lze pozorovat při podávání TMZ v režimu delší dávky. Všichni pacienti léčení TMZ, zejména ti, kteří užívají steroidy, by však měli být pečlivě sledováni kvůli vývoji PCP bez ohledu na dávkovací režim.U pacientů léčených TMZ byly hlášeny případy smrtelného respiračního selhání, zejména v kombinaci s dexamethasonem nebo jinými steroidy.
HBV
Byla hlášena hepatitida způsobená reaktivací viru hepatitidy B (HBV), v některých případech s fatálním koncem. Před zahájením léčby u pacientů s pozitivní sérologií hepatitidy B (včetně pacientů s aktivním onemocněním) je třeba konzultovat odborníky na onemocnění jater. Pacienti by měli být během léčby monitorováni a vhodně léčeni.
Hepatotoxicita
U pacientů léčených TMZ bylo hlášeno poškození jater, včetně smrtelného selhání jater (viz bod 4.8). Před zahájením léčby by měly být provedeny základní testy jaterních funkcí. Pokud jsou výsledky abnormální, měli by lékaři před zahájením TMZ vyhodnotit přínos / riziko včetně možnosti smrtelného selhání jater. U pacientů na 42denním léčebném cyklu by se testy jaterních funkcí měly opakovat v polovině průběhu. U všech pacientů by měly být po každém průběhu léčby provedeny testy jaterních funkcí. U pacientů s významnou poruchou funkce jater by měli lékaři zvážit přínos / riziko pokračování léčby. Jaterní toxicita se může objevit několik týdnů nebo déle po poslední léčbě temozolomidem.
Novotvary
Velmi vzácně byly také hlášeny případy myelodysplastického syndromu a sekundárních malignit, včetně myeloidní leukémie (viz bod 4.8).
Antiemetická terapie
Nevolnost a zvracení jsou u TMZ velmi časté.
Před nebo po podání TMZ může být indikována antiemetická terapie.
Dospělí pacienti s prvním diagnostikovaným multiformním glioblastomem
Antiemetická profylaxe se doporučuje před počáteční dávkou souběžné fáze, zatímco se důrazně doporučuje během fáze monoterapie.
Pacienti s rekurentním nebo progresivním maligním gliomem
U pacientů, u kterých se v předchozích léčebných cyklech vyskytlo závažné zvracení (3. nebo 4. stupně), může být nutná antiemetická léčba.
Laboratorní parametry
U pacientů léčených TMZ se může objevit myelosuprese, včetně prodloužené pancytopenie, což může vést k aplastické anémii, což v některých případech vede k úmrtí. V některých případech hodnocení komplikuje expozice souběžně podávaným léčivým přípravkům spojeným s aplastickou anémií, včetně karbamazepinu, fenytoinu a sulfamethoxazolu / trimethoprimu. Před podáním je třeba vyhodnotit následující laboratorní parametry: ANC ≥ 1,5 x 109 / l a počet krevních destiček ≥ 100 x 109 / l. Kompletní a týdenní krevní obraz by měl být proveden 22. den (21 dní po prvním podání) nebo do 48 hodin, dokud ANC nebude> 1,5 x 109 / l a počet krevních destiček nebude> 100 x 109 / l. Pokud se ANC sníží na krevní destičky, je to 2, 150 mg / m2 a 200 mg / m2. Nejnižší doporučená dávka je 100 mg / m2.
Pediatrická populace
Neexistují žádné klinické zkušenosti s používáním TMZ u dětí mladších 3 let. Klinické zkušenosti u starších dětí a dospívajících jsou velmi omezené (viz body 4.2 a 5.1).
Starší pacienti (> 70 let)
Zdá se, že starší pacienti mají vyšší riziko neutropenie a trombocytopenie než mladší. Proto by měl být TMZ podáván se zvláštní péčí starším pacientům.
Mužští pacienti
Mužům léčeným TMZ by mělo být doporučeno, aby se nerozmnožili do 6 měsíců po poslední dávce a aby se před zahájením léčby informovali o kryokonzervaci spermií (viz bod 4.6).
Laktóza
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
04.5 Interakce s jinými léčivými přípravky a jiné formy interakce
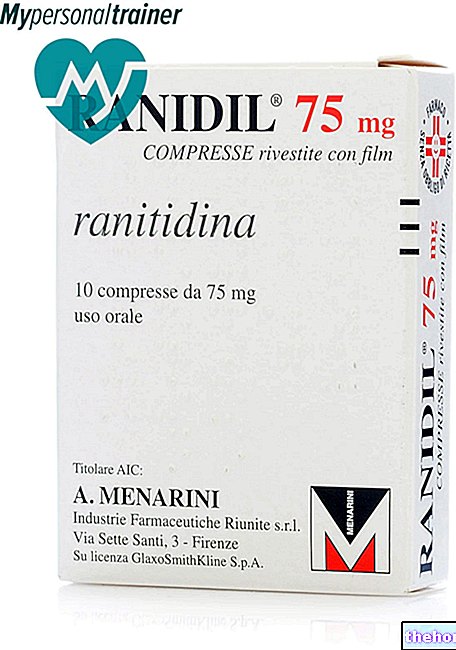
V samostatné studii fáze I podávání TMZ s ranitidinem neměnilo absorpci temozolomidu ani expozici jeho aktivnímu metabolitu monomethyltriazenoimidazol karboxamid (MTIC).
Podávání TMZ s jídlem má za následek 33% pokles Cmax a 9% pokles oblasti pod křivkou (AUC).
Protože nelze vyloučit, že změna Cmax má klinický význam, měl by být přípravek Temodal podáván bez jídla.
Populační farmakokinetické hodnocení studií fáze II ukázalo, že současné podávání dexamethasonu, prochlorperazinu, fenytoinu, karbamazepinu, ondansetronu, antagonistů H2 receptorů nebo fenobarbitalu neměnilo clearance TMZ. Současné podávání kyseliny valproové je spojeno s mírným, ale statisticky významným snížením clearance TMZ.
Nebyly provedeny žádné studie ke stanovení účinku TMZ na metabolismus nebo eliminaci jiných léčivých přípravků. Jelikož však TMZ nepodléhá jaternímu metabolismu a vyznačuje se nízkou vazbou na bílkoviny, je nepravděpodobné, že by to ovlivnilo farmakokinetiku jiných léčivých přípravků ( viz bod 5.2).
Použití TMZ v kombinaci s jinými myelosupresivy může zvýšit možnost myelosuprese.
Pediatrická populace
Interakční studie byly provedeny pouze u dospělých.
04.6 Těhotenství a kojení
Těhotenství
Nejsou k dispozici žádné údaje o těhotných ženách. Teratogenní a / nebo fetální toxicita byla prokázána v preklinických studiích na potkanech a králících léčených 150 mg / m2 TMZ (viz bod 5.3). Temodal by neměl být podáván těhotným ženám. Pokud se zvažuje použití v těhotenství, pacientka by měla být informována o potenciálním riziku pro plod.
Čas krmení
Není známo, zda se TMZ vylučuje do lidského mléka; kojení by proto mělo být během léčby TMZ přerušeno.
Ženy v plodném věku
Ženám ve fertilním věku by mělo být doporučeno používat účinné antikoncepční metody, aby se během léčby TMZ vyhnuly otěhotnění.
Mužská plodnost
TMZ může mít genotoxické účinky. Mužům léčeným TMZ by proto mělo být doporučeno, aby se nerozmnožili do 6 měsíců po poslední dávce a aby se před zahájením léčby informovali o kryokonzervaci spermií kvůli možné nevratné neplodnosti spojené s terapií TMZ.
04.7 Účinky na schopnost řídit a obsluhovat stroje
TMZ má kvůli nástupu únavy a ospalosti minimální vliv na schopnost řídit a obsluhovat stroje (viz bod 4.8).
04.8 Nežádoucí účinky
Zkušenosti z klinických zkoušek
U pacientů léčených TMZ, buď na souběžné léčbě RT nebo jako monoterapie po RT u nově diagnostikovaného multiformního glioblastomu, nebo jako monoterapie u pacientů s relabujícím nebo progresivním gliomem, jsou velmi časté nežádoucí účinky hlášeny podobné a jsou: nauzea, zvracení, zácpa , anorexie, bolest hlavy a únava. Záchvaty byly velmi často hlášeny u pacientů s prvním diagnostikovaným multiformním glioblastomem, kteří dostávali monoterapii, a vyrážka byla velmi často hlášena u pacientů s prvním diagnostikovaným multiformním glioblastomem, kteří dostávali TMZ souběžně s RT a také jako monoterapie a běžně u rekurentního gliomu. Většina hematologických nežádoucích účinků je hlášena jako běžná nebo velmi častá v obou indikacích (tabulky 4 a 5); četnost stupňů 3-4 laboratorních hodnot je uvedena za každou tabulkou.
V tabulkách jsou nežádoucí účinky seřazeny podle třídy orgánových systémů a frekvence. Třídy frekvencí jsou definovány podle následujících konvencí: Velmi časté (≥ 1/10); Časté (≥ 1/100,
Včasná diagnostika multiformního glioblastomu
Tabulka 4 uvádí nežádoucí účinky, které se vyskytly během léčby u pacientů s nově diagnostikovaným multiformním glioblastomem během souběžné a monoterapeutické fáze.
* Jeden pacient, který byl randomizován do ramene pouze RT, obdržel TMZ + RT.
Laboratorní výsledky
Byla pozorována myelosuprese (neutropenie a trombocytopenie), což je známá toxicita omezující dávku u většiny cytotoxických látek, včetně TMZ. Když se laboratorní abnormality sčítají s nežádoucími účinky během souběžné fáze a fáze monoterapie, byly u 8% pacientů pozorovány abnormality neutrofilů 3. nebo 4. stupně včetně neutropenických příhod. Trombocytární změny stupně 3 nebo 4, včetně trombocytopenických příhod, byly pozorovány u 14% pacientů užívajících TMZ.
Rekurentní nebo progresivní maligní gliom
V klinických studiích byly nejčastějšími nežádoucími účinky souvisejícími s léčbou gastrointestinální poruchy, a to nauzea (43%) a zvracení (36%). Tyto příhody byly obvykle stupně 1 nebo 2 (0–5 epizod zvracení za 24 hodin), samy odezněly nebo byly rychle kontrolovány konvenční antiemetickou léčbou. Incidence těžké nevolnosti a zvracení byla 4%.
Tabulka 5 uvádí nežádoucí účinky zjištěné během klinických studií s relabujícím nebo progresivním maligním gliomem a po uvedení Temodalu na trh.
Laboratorní výsledky
Trombocytopenie 3. nebo 4. stupně se vyskytla u 19% a 17% pacientů léčených pro maligní gliom. To vedlo k hospitalizaci a / nebo přerušení léčby TMZ u 8%, respektive 4% pacientů. Myelosuprese byla předvídatelná (obvykle v prvních několika cyklech, s nejnižší hodnotou mezi 21. dnem a 28. dnem) a zotavení bylo rychlé obvykle do 1 roku -2 týdny Nebyl pozorován žádný důkaz kumulativní myelosuprese Přítomnost trombocytopenie může zvýšit riziko krvácení a přítomnost neutropenie nebo leukopenie infekcí.
Sex
V populační farmakokinetické analýze z klinických studií bylo 101 žen a 169 mužských subjektů, pro které byl k dispozici počet nejnižších neutrofilů a 110 ženských a 174 mužských subjektů, pro které byl k dispozici nejnižší počet krevních destiček. Vyšší frekvence neutropenie 4. stupně (ANC vs 5% a byla zjištěna trombocytopenie (vs. 3%, u žen versus mužů, v prvním cyklu terapie). V souboru dat 400 subjektů s rekurentním gliomem se neutropenie 4. stupně vyskytla u 8% žen vs 4% mužů a trombocytopenie stupně 4 u 8% žen vs 3% mužů, v prvním cyklu terapie. Ve studii s 288 subjekty s nově diagnostikovaným multiformním glioblastomem se neutropenie stupně 4 vyskytla u 3% žen vs 0% mužů a trombocytopenie stupně 4 u 1% žen vs 0% mužů, v prvním cyklu terapie.
Pediatrická populace
Orální TMZ byl studován u pediatrických pacientů (ve věku od 3 do 18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v denním dávkovacím režimu po dobu 5 dnů každých 28 dní. Ačkoli jsou údaje omezené, očekává se, že tolerance u dětí bude podobná jako u dospělých. Bezpečnost TMZ u dětí mladších 3 let nebyla stanovena.
Postmarketingové zkušenosti
Během postmarketingové expozice byly zjištěny následující další závažné nežádoucí účinky:
† Včetně případů s fatálním koncem
* Četnosti odhadované na základě příslušných klinických studií.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky, které se vyskytnou po registraci léčivého přípravku, je důležité, protože umožňuje průběžné sledování poměru přínosu a rizika léčivého přípravku. Zdravotničtí pracovníci jsou požádáni, aby hlásili jakékoli podezření na nežádoucí účinky prostřednictvím italské agentury pro léčivé přípravky. , webové stránky: http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Předávkování
Klinicky byly u pacientů klinicky hodnoceny dávky 500, 750, 1 000 a 1 250 mg / m2 (celková dávka na cyklus po dobu 5 dnů). Hematologická toxicita omezovala dávku a byla hlášena u každé dávky, ale očekává se, že bude závažnější při vyšších dávkách. Jeden pacient dostal předávkování 10 000 mg (celková jednorázová dávka po dobu 5 dnů) a hlášené nežádoucí účinky byly pancytopenie, pyrexie, multifunkční selhání a úmrtí. Byly hlášeny případy, kdy pacienti užívali doporučenou dávku déle než 5 dní (až 64 dní), hlásili nežádoucí účinky včetně ablace kostní dřeně, s infekcí nebo bez infekce, v některých případech závažné a prodloužené a s následkem smrti. V případě předávkování je nutné hematologické vyšetření. Podle potřeby by měla být zavedena podpůrná opatření.
05.0 FARMAKOLOGICKÉ VLASTNOSTI
05.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika - jiná alkylační činidla, ATC kód: L01A X03.
Mechanismus účinku
Temozolomide je triazen, který prochází rychlou chemickou přeměnou při fyziologickém pH na účinnou látku monomethyltriazenoimidazol karboxamid (MTIC). Cytotoxicita MTIC je pokládána hlavně za alkylaci v poloze O6 7 guaninu s další alkylací v poloze N. Cytotoxické léze, které se následně vyvinou, jsou považovány za aberantní opravu methylovaného aduktu.
Klinická účinnost a bezpečnost
Včasná diagnostika multiformního glioblastomu
Celkem 573 pacientů bylo randomizováno tak, aby dostávali buď TMZ + RT (n = 287), nebo samotný RT (n = 286). Pacienti v rameni TMZ + RT dostávali souběžně TMZ (75 mg / m monoterapie TMZ (150 - 200 mg / m) ve dnech 1 - 5 každého 28denního cyklu, až do maximálně 6 cyklů, počínaje 4 týdny po ukončení RT. kontrolní rameno dostalo pouze RT. Profylaxe proti zápalu plic byla během RT a kombinované terapie s TMZ. Pneumocystis jirovecii (PCP).
TMZ byl podáván jako záchranná terapie v následné fázi u 161 pacientů z 282 (57%) v rameni samotném RT a u 62 pacientů z 277 (22%) v rameni TMZ + RT.
L"úroveň ohrožení (HR) pro celkové přežití bylo 1,59 (95% CI pro HR = 1,33 - 1,91) s log -rank p vs 10%) je vyšší v rameni RT + TMZ. Přidání souběžného TMZ k RT, následované samotným TMZ, při léčbě pacientů s nově diagnostikovaným multiformním glioblastomem, prokázalo statisticky významné zvýšení celkového přežití (OS) ve srovnání s RT samotným.
Výsledky studie nebyly konzistentní v podskupině pacientů s nízkým výkonem (WHO PS = 2, n = 70), u nichž bylo celkové přežití a doba do progrese v obou ramenech podobné. Zdá se však, že v této skupině pacientů není přípustná míra rizika.
Rekurentní nebo progresivní maligní gliom
Údaje o klinické účinnosti u pacientů s progresivním nebo relapsujícím multiformním glioblastomem (Karnofsky výkonnostní stav [KPS] ≥ 70) po chirurgickém zákroku a RT byly získány ve dvou klinických studiích s orálním TMZ. Jeden provedený na 138 pacientech (29% z nich dříve podstoupilo chemoterapii) byl nekomparativní a druhý proveden pomocí TMZ vs prokarbazin u 225 pacientů (67% z nich dříve podstoupilo chemoterapii na bázi nitrosomočoviny) bylo randomizováno do aktivní kontroly. V obou studiích bylo primárním cílovým parametrem přežití bez progrese (PFS) definované MRI nebo neurologické zhoršení. V nekomparativní studii bylo 6měsíční PFS 19%, medián přežití bez progrese byl 2,1 měsíce a medián celkového přežití bylo 5,4 měsíce Incidence objektivní odpovědi (ORR) na základě MRI byla 8%.
V randomizované, aktivně kontrolované studii byl PFS po 6 měsících významně vyšší u TMZ než u prokarbazinu (21% vs. 8%, v tomto pořadí-chi-square p = 0,008) s mediánem PFS 2,89, respektive 1,88 měsíce ( log rank test p = 0,0063). Medián přežití TMZ a prokarbazinu byl 7,34, respektive 5,66 měsíce (log rank test p = 0,33). Po 6 měsících bylo procento přeživších pacientů významně vyšší v rameni TMZ (60%) než v rameni s prokarbazinem (44%) (chí-kvadrát p = 0,019). Přínos byl pozorován u dříve podstupujících pacientů s chemoterapií s KPS ≥ 80.
Údaje o čase do zhoršení neurologického stavu byly pro TMZ příznivé ve srovnání s prokarbazinem, stejně jako údaje o čase do zhoršení výkonnostního stavu (pokles KPS při
Opakující se anaplastický astrocytom
V multicentrické prospektivní studii fáze II hodnotící bezpečnost a účinnost orálního TMZ při léčbě pacientů s anaplastickým astrocytomem při prvním relapsu činil 6měsíční PFS 46%. Medián PFS byl 5,4 měsíce. Medián celkového přežití byla 14,6 měsíce. Míra odpovědi na základě hodnocení centrálního recenzenta byla 35% (13 CR a 43 PR) u populace určené k léčbě (ITT) n = 162 U 43 pacientů bylo hlášeno stabilní onemocnění. volné přežití pro populaci ITT bylo 44% s mediánem přežití bez příhod 4,6 měsíce; tyto výsledky jsou podobné těm pro přežití bez progrese. U populace vhodné pro histologii byly výsledky účinnosti podobné. Dosažení objektivního radiologického odezva nebo údržba bez progrese byla silně spojena s údržbou resp zlepšení kvality života.
Pediatrická populace
Orální TMZ byl studován u pediatrických pacientů (ve věku od 3 do 18 let) s rekurentním gliomem mozkového kmene nebo rekurentním astrocytomem vysokého stupně v denním dávkovacím režimu po dobu 5 dnů každých 28 dní. Tolerance vůči TMZ byla u dospělých podobná.
05.2 Farmakokinetické vlastnosti
TMZ je spontánně hydrolyzován při fyziologickém pH primárně v aktivní formě, 3-methyl- (triazen-1-yl) imidazol-4-karboxamidu (MTIC). MTIC se spontánně hydrolyzuje na 5-aminoimidazol-4-karboxamid (AIC), známý meziprodukt v biosyntéze purinů a nukleových kyselin, a na methylhydrazin, o kterém se věří, že je aktivní alkylační formou. MTIC je primárně způsobeno alkylací DNA hlavně v polohách O6 7 a N guaninu. Pokud jde o AUC TMZ, expozice MTIC a AIC je 2,4%, respektive 23%. In vivo, t1 / 2 MTIC byl podobný jako u TMZ a roven 1,8 h.
Vstřebávání
Po orálním podání u dospělých pacientů se TMZ rychle vstřebává a vrcholových koncentrací je dosaženo již za 20 minut po podání dávky (průměrná doba mezi 0,5 a 1,5 hodinou). Po orálním podání TMZ značené 1414C bylo průměrné vylučování C stolicí po dobu 7 dnů po dávce 0,8%, což dokazuje úplnou absorpci.
Rozdělení
TMZ se vyznačuje nízkou tendencí vázat se na proteiny (10% až 20%), a proto se neočekává, že bude interagovat s látkami, které se silně váží na proteiny.
Studie PET na lidech a preklinické údaje naznačují, že TMZ rychle překračuje hematoencefalickou bariéru a je přítomen v CSF. Penetrace CSF byla potvrzena u jednoho pacienta; expozice CSF vypočtená na základě AUC TMZ byla přibližně 30% expozice plazmy, což bylo v souladu s údaji o zvířatech.
Odstranění
Plazmatický poločas (t1 / 2) v plazmě je přibližně 1,8 hodiny. Hlavní cestou eliminace 14C je ledvina. Po orálním podání se přibližně 5% - 10% dávky v nezměněné podobě získá v moči za 24 hodin. hodiny a zbytek se vyloučí jako kyselina temozolomidová, 5-aminoimidazol-4-karboxamid (AIC) nebo jako neidentifikované polární metabolity.
Plazmatické koncentrace se zvyšují způsobem závislým na dávce. Plazmatická clearance, distribuční objem a poločas jsou nezávislé na dávce.
Zvláštní populace
Populační farmakokinetická analýza ukázala, že plazmatická clearance TMZ byla nezávislá na věku, renálních funkcích a užívání tabáku.V oddělené farmakokinetické studii byly plazmatické farmakokinetické profily u pacientů s mírnou poruchou funkce jater až středně závažnou podobné profilům pozorovaným u pacientů s normální funkcí jater.
Dětští pacienti měli vyšší AUC než dospělí pacienti; maximální tolerovaná dávka (MDT) však byla 1 000 mg / m2 za cyklus u dětí i dospělých.
05.3 Předklinické údaje vztahující se k bezpečnosti
Byly provedeny jednocyklové studie toxicity (5 dní léčby a 23 dní bez léčby), 3 a 6 cyklů na potkanech a psech.Primární cíle toxicity zahrnovaly kostní dřeň, lymfoetikulární systém, varlata a gastrointestinální trakt a při vyšších dávkách, které byly smrtelné u 60% -100% testovaných potkanů a psů, došlo k degeneraci sítnice. Většina toxických účinků byla reverzibilní, kromě nežádoucích účinků ovlivňujících mužský reprodukční systém a degenerace sítnice. Protože jsou však dávky vedoucí k degeneraci sítnice v rozmezí smrtelných dávek a v klinických studiích nebyly pozorovány žádné srovnatelné účinky, nebyla tomuto zjištění přisuzována žádná klinická relevance.
TMZ je embryotoxické, teratogenní a genotoxické alkylační činidlo. TMZ je toxičtější u potkanů a psů než u lidí a klinická dávka se blíží nejnižší smrtelné dávce pro krysy a psy. Snížení počtu leukocytů a krevních destiček závislé na dávce se zdá být významným indikátorem toxicity. Ve studii se 6 studiemi V krysích cyklech byly pozorovány různé novotvary, včetně karcinomu prsu, kožního keratoakantomu, adenomu bazálních buněk, zatímco ve studiích na psech nebyly pozorovány žádné nádory ani pre-neoplastické změny. Krysy jsou zvláště citlivé na onkogenní účinky TMZ. první nádory se objevují do 3 měsíců od začátku podávání. Tato doba latence je také velmi krátká pro alkylační činidlo.
Výsledky testu Ames / Salmonella a testu chromozomové aberace lymfocytů lidské periferní krve (HPBL) ukázaly pozitivní odpověď mutagenity.
06.0 FARMACEUTICKÉ INFORMACE
06.1 Pomocné látky
Obsah tobolky:
bezvodá laktóza,
koloidní bezvodý oxid křemičitý,
sodná sůl karboxymetylškrobu typu A,
kyselina vinná,
kyselina stearová.
Obal tobolky:
želé,
oxid titaničitý (E 171),
laurylsulfát sodný,
červený oxid železitý (E 172)
Tiskový inkoust:
šelak,
propylenglykol,
čištěná voda,
hydroxid amonný,
hydroxid draselný,
černý oxid železitý (E 172).
06.2 Neslučitelnost
Irelevantní.
06.3 Doba platnosti
3 roky
06.4 Zvláštní opatření pro skladování
Prezentace lahví
Uchovávejte při teplotě do 30 ° C.
Tobolky uchovávejte v původní lahvičce, aby byly chráněny před vlhkostí.
Láhev uchovávejte těsně uzavřenou.
Prezentace ve sáčku
Uchovávejte při teplotě do 30 ° C.
06.5 Charakter vnitřního obalu a obsah balení
Prezentace lahví
Láhve z tmavého skla typu I s dětským bezpečnostním polypropylenovým uzávěrem obsahující 5 nebo 20 tvrdých tobolek.
Krabice obsahuje jednu láhev.
Prezentace ve sáčku
Sáčky jsou složeny z lineárního polyetylenu s nízkou hustotou (nejvnitřnější vrstva), hliníku nebo polyethylentereftalátu.
Každý sáček obsahuje 1 tvrdou tobolku a je dodáván v papírové krabičce.
Karton obsahuje 5 nebo 20 tvrdých tobolek v jednotlivě uzavřených sáčcích.
Na trhu nemusí být všechny velikosti balení.
06.6 Návod k použití a zacházení
Neotevírejte tobolky. Pokud je tobolka poškozená, vyvarujte se kontaktu prášku v ní obsaženého s kůží nebo sliznicemi. Pokud dojde ke kontaktu s pokožkou nebo sliznicemi, okamžitě a důkladně omyjte postižené místo mýdlem a vodou.
Pacienti by měli být poučeni, aby kapsle uchovávali mimo dohled a dosah dětí, nejlépe v uzamčené skříňce. Náhodné požití může být pro děti smrtelné.
Nepoužitý léčivý přípravek a odpad z tohoto přípravku musí být zlikvidován v souladu s místními předpisy.
07.0 DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited
Hertford Road, Hoddesdon
Hertfordshire EN11 9BU
Spojené království
08.0 REGISTRAČNÍ ČÍSLO
EU/1/98/096/005
EU/1/98/096/006
EU/1/98/096/015
EU/1/98/096/016
034527059
034527061
034527150
034527162
09.0 DATUM PRVNÍ REGISTRACE NEBO PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. ledna 1999
Datum posledního obnovení: 26. ledna 2009
10.0 DATUM REVIZE TEXTU
27. května 2015








.jpg)