Účinné látky: Rivaroxaban
Xarelto 15 mg potahované tablety
Xarelto 20 mg potahované tablety
Příbalové informace Xarelto jsou k dispozici pro velikosti balení: - Xarelto 2,5 mg potahované tablety
- Xarelto 10 mg potahované tablety
- Xarelto 15 mg potahované tablety, Xarelto 20 mg potahované tablety
Proč se používá Xarelto? K čemu to je?
Xarelto obsahuje léčivou látku rivaroxaban a používá se u dospělých k
- zabraňte tvorbě krevních sraženin v mozku (mrtvice) a jiných cévách v těle, pokud máte typ nepravidelného srdečního rytmu nazývaný nevalvulární fibrilace síní.
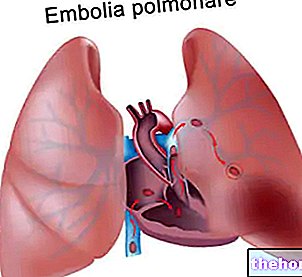
- léčit krevní sraženiny v žilách nohou (hluboká žilní trombóza) a v cévách plic (plicní embolie) a zabránit návratu krevních sraženin do cév nohou a / nebo plic.
Xarelto patří do skupiny léků nazývaných antitrombotika. Jeho působení je způsobeno blokováním srážecího faktoru (faktor Xa), po kterém následuje snížená tendence krve vytvářet sraženiny.
Kontraindikace Kdy by neměl být Xarelto používán
Neužívejte Xarelto
- jestliže jste alergický (á) na rivaroxaban nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže máte nadměrné krvácení (krvácení)
- pokud máte v části těla onemocnění nebo stav, který zvyšuje riziko závažného krvácení (např. žaludeční vředy, rány nebo krvácení do mozku, nedávná operace mozku nebo očí)
- jestliže užíváte léky k prevenci srážení (např. warfarin, dabigatran, apixaban nebo hepariny), kromě případů, kdy změníte antikoagulační terapii nebo když dostáváte hepariny venózním nebo arteriálním katetrem, aby byl otevřený.
- pokud máte onemocnění jater, které zvyšuje riziko krvácení,
- během těhotenství nebo kojení
Neužívejte přípravek Xarelto a informujte svého lékaře, pokud se vás týká některý z popsaných stavů.
Opatření pro použití Co potřebujete vědět, než začnete Xarelto užívat
Před užitím přípravku Xarelto se poraďte se svým lékařem nebo lékárníkem
Zvláštní pozornost věnujte přípravku Xarelto
- pokud máte zvýšené riziko krvácení, což by mohlo být, pokud máte:
- závažné onemocnění ledvin, protože funkce ledvin může změnit množství léčiva, které je v těle aktivní
- jestliže užíváte jiné léky k prevenci srážení (např. warfarin, dabigatranetexilát, apixaban nebo heparin), pokud měníte antikoagulační léčbu nebo když dostáváte heparin venózním nebo arteriálním katetrem, aby byl otevřený (viz bod „Další léčivé přípravky a přípravek Xarelto ")
- poruchy krvácení
- velmi vysoký krevní tlak, nekontrolovaný léky
- onemocnění žaludku nebo střev, která by mohla způsobit krvácení, jako je zánět střev nebo žaludku nebo zánět jícnu, například způsobený gastroezofageálním refluxem (onemocnění, při kterém kyselost žaludku stoupá do jícnu)
- porucha krevních cév v zadní části oka (retinopatie)
- plicní onemocnění se zvětšenými hnisavými průduškami (bronchiektázie) nebo předchozí krvácení z plic
- nádor lokalizovaný v kritickém orgánu těla
- pokud máte protetickou srdeční chlopeň
- pokud lékař zjistí, že je váš krevní tlak nestabilní nebo je plánována jiná léčba nebo chirurgický zákrok k odstranění krevních sraženin z plic
Pokud se vás týká cokoli z výše uvedeného, sdělte to svému lékaři dříve, než začnete přípravek Xarelto užívat. Váš lékař rozhodne, zda byste měli být léčeni tímto léčivým přípravkem a zda byste měli být pečlivě sledováni.
Pokud potřebujete operaci:
- Je velmi důležité, abyste užíval (a) přípravek Xarelto před a po operaci přesně v časech uvedených lékařem.
Děti a dospívající
Přípravek Xarelto se nedoporučuje osobám mladším 18 let. O jeho použití u dětí a dospívajících nejsou dostatečné informace.
Interakce Které léky nebo potraviny mohou změnit účinek přípravku Xarelto
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval (a) nebo které možná budete užívat, a to io lécích, které jsou dostupné bez lékařského předpisu.
- Pokud užíváte:
- některé léky na mykózy (např. ketokonazol, itrakonazol, vorikonazol, posakonazol), pokud nejsou aplikovány pouze na kůži
- některá antivirotika pro léčbu HIV / AIDS (např. ritonavir)
- jiné léky používané k inhibici srážení (např. enoxaparin, klopidogrel nebo antagonisté vitaminu K, jako je warfarin a acenokumarol)
- léky působící proti zánětům a bolesti (např. naproxen nebo kyselina acetylsalicylová)
- dronedaron, lék používaný k léčbě fibrilace síní
Pokud se vás týká některý z popsaných stavů, sdělte to svému lékaři před užitím přípravku Xarelto, protože účinek přípravku Xarelto se může zvýšit.Lékař rozhodne, zda byste měli být léčeni tímto přípravkem a zda byste měli být pečlivě sledováni.
Pokud si váš lékař myslí, že máte zvýšené riziko vzniku žaludečních nebo střevních vředů, může vám předepsat preventivní léčbu vředů.
- Pokud užíváte:
- některé léky k léčbě epilepsie (fenytoin, karbamazepin, fenobarbital)
- Třezalka tečkovaná (Hypericum perforatum), bylinný přípravek používaný k léčbě deprese
- rifampicin, antibiotikum
Pokud se vás týká cokoli z výše uvedeného, sdělte to svému lékaři před užitím přípravku Xarelto, protože účinky přípravku Xarelto mohou být sníženy. Váš lékař rozhodne, zda byste měli být léčeni přípravkem Xarelto a zda byste měli být pečlivě sledováni.
Varování Je důležité vědět, že:
Těhotenství a kojení
Neužívejte přípravek Xarelto, pokud jste těhotná nebo kojíte. Pokud existuje možnost otěhotnění, používejte při užívání přípravku Xarelto spolehlivou metodu antikoncepce. Pokud otěhotníte během užívání tohoto léku, okamžitě to sdělte svému lékaři, který rozhodne, jak pokračovat v léčbě.
Řízení dopravních prostředků a obsluha strojů
Xarelto může způsobit závratě (častý nežádoucí účinek) nebo mdloby (méně častý nežádoucí účinek) (viz bod 4 „Možné nežádoucí účinky“). Pokud se tyto příznaky objeví, neřiďte ani neobsluhujte stroje.
Xarelto obsahuje laktózu.
Pokud vám lékař řekl, že trpíte „nesnášenlivostí některých cukrů, kontaktujte svého lékaře před užitím tohoto léčivého přípravku.
Dávka, způsob a doba podání Jak používat Xarelto: Dávkování
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud máte pochybnosti, poraďte se se svým lékařem nebo lékárníkem.
Jakou dávku užít
- Prevence krevních sraženin v mozku (mrtvice) a jiných cév v těle Doporučená dávka je jedna 20mg tableta jednou denně. Pokud máte problémy s ledvinami, lze dávku snížit na jednu 15mg tabletu jednou denně. Jednou den.
- K léčbě krevních sraženin v žilách nohou a v cévách plic a k zabránění návratu sraženin. Doporučená dávka je jedna 15mg tableta dvakrát denně po dobu prvních 3 týdnů. Po 3 týdnech je doporučená dávka jedna 20mg tableta jednou denně.Pokud máte problémy s ledvinami, může se lékař rozhodnout po 3 týdnech snížit dávku léčby na jednu 15mg tabletu jednou denně, pokud je riziko krvácení větší než riziko vzniku nové krevní sraženiny.
Tabletu nebo tablety spolkněte nejlépe s trochou vody. Užívejte Xarelto s jídlem.
Pokud máte potíže s polykáním celé tablety, zeptejte se svého lékaře, jak jinak Xarelto užívat. Tableta může být rozdrcena a smíchána s trochou vody nebo jablečného pyré bezprostředně před užitím.
V případě potřeby vám lékař může dát rozdrcenou tabletu Xarelto hadičkou zavedenou do žaludku.
Kdy užívat Xarelto
Tablety užívejte každý den, dokud vám lékař neřekne, abyste přestali.
Snažte se vždy užívat tabletu (y) ve stejnou denní dobu, abyste si snáze pamatovali.
Lékař určí délku léčby.
Předávkování Co dělat, když jste užil příliš mnoho přípravku Xarelto
Jestliže jste užil (a) více přípravku Xarelto, než jste měl (a)
Pokud jste užil příliš mnoho tablet Xarelto, okamžitě kontaktujte svého lékaře. Pokud jste užili příliš mnoho přípravku Xarelto, zvyšuje se riziko krvácení.
Jestliže jste zapomněl (a) užít Xarelto
- Pokud užíváte jednu 20mg tabletu nebo jednu 15mg tabletu jednou denně a zapomněli jste dávku, užijte ji, jakmile si vzpomenete. Neužívejte více než jednu tabletu ve stejný den, abyste nahradili zapomenutou dávku. Další tabletu užijte následující den a poté pokračujte v užívání jedné tablety jednou denně.
- Pokud užíváte jednu 15mg tabletu dvakrát denně a zapomněli jste dávku, užijte ji, jakmile si vzpomenete. Neužívejte více než dvě 15mg tablety v jeden den. Pokud zapomenete dávku, můžete užít dvě 15mg tablety současně, abyste v jeden den užili celkem dvě tablety (30 mg). Další den pokračujte jednou 15mg tabletou dvakrát denně.
Jestliže jste přestal (a) užívat přípravek Xarelto
Nepřestávejte užívat Xarelto, aniž byste si to nejprve promluvili se svým lékařem, protože Xarelto léčí vážná onemocnění a brání jejich vzniku.
Máte -li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Nežádoucí účinky Jaké jsou vedlejší účinky přípravku Xarelto
Podobně jako všechny léky, může mít i Xarelto nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Stejně jako ostatní podobné léky (antitrombotika) může Xarelto způsobit potenciálně život ohrožující krvácení. Masivní krvácení může způsobit náhlý pokles krevního tlaku (šok). V některých případech nemusí dojít ke krvácení. Je zřejmé.
Možné nežádoucí účinky, které mohou naznačovat krvácení:
Okamžitě informujte svého lékaře, pokud zaznamenáte některý z následujících nežádoucích účinků:
- dlouhodobá nebo nadměrná ztráta krve
- neobvyklá slabost, únava, bledá kůže, závratě, bolesti hlavy, otoky neznámého původu, dušnost, bolest na hrudi nebo angina pectoris, což mohou být příznaky krvácení,
Váš lékař se může rozhodnout vás pečlivě sledovat nebo změnit typ léčby.
Celkový seznam možných vedlejších účinků:
Časté (mohou postihnout až 1 z 10 uživatelů)
- krvácení do žaludku nebo střev, urogenitální krvácení (včetně krve v moči a silné menstruace), krvácení z nosu, gingivální krvácení
- krvácení do oka (včetně krvácení do očního bělma)
- krvácení do tkání nebo tělesné dutiny (hematom, podlitiny)
- vykašlávání krve
- krvácení z kůže nebo pod kůži
- krvácení po operaci
- ztráta krve nebo tekutiny z operační rány
- otoky končetin
- bolest v končetinách
- horečka
- snížení počtu červených krvinek, což může způsobit bledost a slabost nebo dušnost
- bolest žaludku, poruchy trávení, nevolnost nebo zvracení, zácpa, průjem
- nízký krevní tlak (příznaky zahrnují závratě nebo mdloby ve stoje)
- snížená síla a energie (slabost, únava), bolest hlavy, závratě,
- vyrážka, svědění
- porucha funkce ledvin (to lze zjistit testy provedenými lékařem)
- zvýšení některých jaterních enzymů v krevních testech
Méně časté (mohou postihnout až 1 ze 100 uživatelů)
- krvácení do mozku nebo uvnitř lebky
- krvácení v jednom kloubu, způsobující bolest a otok
- mdloby
- nevolnost
- suchá ústa
- rychlý srdeční tep
- alergické reakce, včetně alergických kožních reakcí
- kopřivka
- porucha funkce jater (to lze zjistit testy provedenými lékařem)
- krevní testy mohou ukázat zvýšení bilirubinu, některých enzymů ve slinivce nebo játrech nebo počtu krevních destiček
Vzácné (mohou postihnout až 1 z 1000 uživatelů)
- krvácení do svalu
- lokalizovaný otok
- zežloutnutí kůže a očí (žloutenka)
- tvorba krve (hematom) ve slabinách jako komplikace srdečního výkonu, který zahrnuje zavedení katétru do tepny nohy (pseudoaneuryzma)
Frekvence není známa (frekvenci nelze z dostupných údajů určit)
- zvýšený tlak ve svalech nohou nebo paží po „krvácení“, které způsobuje bolest, otok, změny citlivosti, necitlivost nebo paralýzu (kompartment syndrom po „krvácení“)
- poškození ledvin po těžkém krvácení
Od registrace léčivého přípravku byly pozorovány následující nežádoucí účinky: angioedém a alergický edém (otok obličeje, rtů, úst, jazyka nebo hrdla).
Hlášení nežádoucích účinků
Pokud se u vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Nežádoucí účinky můžete hlásit také přímo na adresu: Státní ústav pro kontrolu léčiv Šrobárova 48100 41 Praha 10 Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
Expirace a retence
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na každém blistru za EXP / EXP. Doba použitelnosti se vztahuje k poslednímu dni daného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Pomůže to chránit životní prostředí.
Složení a léková forma
Co přípravek Xarelto obsahuje
- Léčivou látkou je rivaroxaban. Jedna tableta obsahuje rivaroxaban 15 mg nebo 20 mg.
- Dalšími složkami jsou:
Jádro tablety: mikrokrystalická celulóza, sodná sůl kroskarmelózy, monohydrát laktózy, hypromelóza, laurylsulfát sodný, stearát hořečnatý.
Potah tablety: makrogol 3350, hypromelosa, oxid titaničitý (E 171), červený oxid železitý (E 172).
Jak Xarelto vypadá a obsah balení
Xarelto 15 mg potahované tablety jsou červené, kulaté, bikonvexní, na jedné straně s vyraženým křížem BAYER a na druhé straně s vyraženým „15“ a trojúhelníkem.
Tablety jsou dodávány v blistrech v krabičkách po 14, 28, 42 nebo 98 potahovaných tabletách nebo perforovaných jednodávkových blistrech v krabičkách po 10 x 1 nebo 100 x 1 potahovaných tabletách nebo ve více baleních obsahujících 10 balení, z nichž každé obsahuje 10 x 1 potahované tablety.
Xarelto 20 mg potahované tablety jsou červenohnědé, kulaté, bikonvexní, na jedné straně s vyraženým křížem BAYER a na druhé straně s vyraženým „20“ a trojúhelníkem.
Tablety jsou dodávány v blistrech v krabičkách po 14, 28 nebo 98 potahovaných tabletách nebo v perforovaných jednodávkových blistrech v krabičkách po 10 x 1 nebo 100 x 1 potahovaných tabletách nebo ve více baleních obsahujících 10 balení, z nichž každé obsahuje 10 x 1 film. -potahované tablety.
Na trhu nemusí být všechny velikosti balení.
Zdroj příbalové informace: AIFA (Italská agentura pro léčivé přípravky). Obsah zveřejněný v lednu 2016. Přítomné informace nemusí být aktuální.
Abyste měli přístup k nejaktuálnější verzi, doporučujeme navštívit webovou stránku AIFA (Italská agentura pro léčivé přípravky). Prohlášení a užitečné informace.
01.0 NÁZEV LÉČIVÉHO PŘÍPRAVKU
TABLETY XARELTO 20 MG potažené filmem
▼ Léčivý přípravek podléhá dalšímu sledování. To umožní rychlou identifikaci nových bezpečnostních informací. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky. Informace o hlášení nežádoucích účinků viz bod 4.8.
02.0 KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje rivaroxaban 20 mg.
Pomocná látka se známými účinky:
jedna potahovaná tableta obsahuje 21,76 mg laktózy (jako monohydrát), viz bod 4.4.
Úplný seznam pomocných látek viz bod 6.1.
03.0 LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Kulaté, bikonvexní, červenohnědé tablety (průměr 6 mm, poloměr zakřivení 9 mm), na jedné straně vyražený kříž BAYER a na druhé straně „20“ a trojúhelník.
04.0 KLINICKÉ INFORMACE
04.1 Terapeutické indikace
Prevence cévní mozkové příhody a systémové embolie u dospělých pacientů s nevalvulární fibrilací síní s jedním nebo více rizikovými faktory, jako je městnavé srdeční selhání, hypertenze, věk ≥ 75 let, diabetes mellitus, předchozí cévní mozková příhoda nebo přechodný ischemický záchvat.
Léčba hluboké žilní trombózy (DVT) a plicní embolie (PE) a prevence recidivy DVT a PE u dospělých. (viz bod 4.4 pro hemodynamicky nestabilní pacienty s PE)
04.2 Dávkování a způsob podání
Dávkování
Prevence mrtvice a systémové embolie
Doporučená dávka je 20 mg jednou denně a je maximální doporučenou dávkou.
Léčba přípravkem Xarelto by měla pokračovat dlouhodobě, pokud přínos prevence cévní mozkové příhody a systémové embolie převáží riziko krvácení (viz bod 4.4).
Pokud dojde k vynechání dávky, pacient by měl okamžitě užít Xarelto a pokračovat další den s doporučeným dávkováním jednou denně. Ve stejný den by neměla být podána dvojnásobná dávka, aby se nahradila zapomenutá dávka.
Léčba DVT, léčba PE a prevence recidivy DVT a PE
Doporučená dávka pro počáteční léčbu akutní DVT nebo PE je 15 mg dvakrát denně po dobu prvních tří týdnů, poté 20 mg jednou denně pro pokračující léčbu a prevenci recidivy DVT a EP, jak je uvedeno v tabulce níže.
Trvání léčby by mělo být individualizováno po „pečlivém zhodnocení přínosu léčby ve vztahu k riziku krvácení (viz bod 4.4). Krátké trvání léčby (nejméně 3 měsíce) by mělo“ být založeno na přechodných rizikových faktorech (např. nedávná operace, stejně jako trauma nebo „imobilizace), přičemž delší doba musí být založena na trvalých rizikových faktorech nebo idiopatické DVT nebo PE.
Pokud ve fázi léčby vynecháte dávku 15 mg dvakrát denně (den 1 - 21), pacient by měl okamžitě užít Xarelto, aby zajistil denní příjem Xarelta 30 mg. V tomto případě je lze užít. Dva 15 mg tablety ve stejnou dobu. Následující den by měl pacient pokračovat v obvyklém doporučeném příjmu 15 mg dvakrát denně.
Pokud dojde k vynechání dávky ve fázi léčby jednou denně (22. den a později), měl by pacient okamžitě užít přípravek Xarelto a pokračovat další den s doporučeným dávkováním jednou denně. Ve stejný den by neměla být podána dvojnásobná dávka … doplnit zapomenutou dávku.
Přechod z antagonistů vitaminu K (AVK) na Xarelto
U pacientů podstupujících léčbu k prevenci cévní mozkové příhody a systémové embolie by měla být léčba VKA přerušena a zahájena léčba přípravkem Xarelto, když je mezinárodní normalizovaný poměr (INR) ≤ 3,0.
U pacientů podstupujících léčbu DVT, PE a prevenci relapsu by měla být léčba VKA přerušena a léčba Xareltem zahájena, když je INR ≤ 2,5.
U pacientů přecházejících z VKA na Xarelto budou hodnoty INR po podání přípravku Xarelto falešně zvýšeny. INR není určen k měření antikoagulační aktivity přípravku Xarelto, a proto by neměl být používán (viz bod 4.5).
Přechod z Xarelta na antagonisty vitaminu K (AVK)
Během přechodu z Xarelta na AVK existuje možnost neadekvátního antikoagulačního účinku. Kdykoli je provedena změna jiného antikoagulancia, musí být zajištěna adekvátní a kontinuální úroveň antikoagulace. Všimněte si, že Xarelto může pomoci zvýšit INR.
U pacientů přecházejících z Xarelta na VKA by měly být VKA podávány v kombinaci, dokud INR není ≥ 2,0. V prvních dvou dnech přechodné fáze by měla být počáteční standardní dávkou dávka VKA a poté bude vycházet z „INR“. Ve fázi souběžné léčby přípravkem Xarelto a AVK by měl být INR stanoven nejdříve 24 hodin po předchozí dávce přípravku Xarelto, ale před další dávkou. Po přerušení léčby přípravkem Xarelto lze podle toho stanovit INR. Spolehlivě po nejméně 24 od poslední dávky uplynuly hodiny (viz body 4.5 a 5.2).
Přechod z parenterálních antikoagulancií na Xarelto
U pacientů léčených parenterálním antikoagulantem přerušte léčbu parenterálním antikoagulantem a zahajte léčbu přípravkem Xarelto 0 až 2 hodiny před časem, kdy má následovat další parenterální léčivý přípravek (např. Nízký hmotnostní heparin). Molekulární) nebo po vysazení kontinuálního parenterální léčivý přípravek (např. intravenózní nefrakcionovaný heparin).
Přechod z přípravku Xarelto na parenterální antikoagulancia
Podejte první dávku parenterálního antikoagulancia, když měla být podána další dávka přípravku Xarelto.
Zvláštní populace
Porucha funkce ledvin
Omezené klinické údaje u pacientů s těžkou poruchou funkce ledvin (clearance kreatininu 15 - 29 ml / min) naznačují, že plazmatické koncentrace rivaroxabanu jsou významně zvýšeny. Proto by měl být přípravek Xarelto u těchto pacientů používán s opatrností. Použití u pacientů s clearance kreatininu se nedoporučuje
U pacientů se středně těžkou (clearance kreatininu 30 - 49 ml / min) nebo těžkou (clearance kreatininu 15 - 29 ml / min) poruchou funkce ledvin platí následující doporučení dávkování:
• Pro prevenci cévní mozkové příhody a systémové embolie u pacientů s nevalvulární fibrilací síní je doporučená dávka 15 mg jednou denně (viz bod 5.2).
• pro léčbu DVT, léčbu PE a prevenci recidivy DVT a PE: pacienti by měli být léčeni 15 mg dvakrát denně po dobu prvních 3 týdnů. Poté je doporučená dávka 20 mg jednou denně. Snížení dávky z 20 mg jednou denně na 15 mg jednou denně by mělo být zváženo pouze tehdy, pokud je hodnocení rizika krvácení u pacienta větší než riziko recidivy HŽT a PE. Doporučení pro použití 15 mg je založeno na farmakokinetickém modelování a v klinickém prostředí nebylo studováno (viz body 4.4, 5.1 a 5.2).
U pacientů s mírnou poruchou funkce ledvin (clearance kreatininu 50 - 80 ml / min) není nutná úprava dávky (viz bod 5.2).
Porucha funkce jater
Xarelto je kontraindikován u pacientů s jaterním onemocněním spojeným s koagulopatií a klinicky významným rizikem krvácení, včetně pacientů s cirhózou a Child Pugh B a C (viz body 4.3 a 5.2).
Starší populace
Žádná úprava dávky (viz bod 5.2).
Tělesná hmotnost
Žádná úprava dávky (viz bod 5.2).
Sex
Žádná úprava dávky (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Xarelto u dětí ve věku 0-18 let nebyla stanovena. Vzhledem k nedostatku údajů se podávání přípravku Xarelto nedoporučuje u dětí mladších 18 let.
Pacienti podstupující kardioverzi
Léčbu přípravkem Xarelto lze zahájit nebo v ní pokračovat u pacientů vyžadujících kardioverzi.
U kardioverze vedené transesofageálním ultrazvukem (TEE) u pacientů, kteří nebyli dříve léčeni antikoagulancii, by měla být léčba přípravkem Xarelto zahájena nejméně 4 hodiny před kardioverzí, aby byla zajištěna adekvátní antikoagulace (viz body 5.1 a 5.2). U všech pacientů musíte potvrdit že přípravek Xarelto byl užíván předepsaným způsobem před zahájením kardioverze. Rozhodnutí o zahájení a délce léčby by měla být přijata s přihlédnutím k doporučením oficiálních pokynů pro antikoagulační léčbu u pacientů podstupujících kardioverzi.
Způsob podání
K perorálnímu podání.
Tablety se užívají s jídlem (viz bod 5.2).
U pacientů, kteří nemohou polykat tablety celé, lze tabletu Xarelto rozdrtit a smíchat s trochou vody nebo jablečného pyré bezprostředně před použitím a podávat orálně. Po podání rozdrcených potahovaných tablet 15 mg nebo 20 mg přípravku Xarelto by měla dávka hned následovat jídlo.
Jakmile je tableta Xarelto rozdrcena, lze ji také podávat sondou, s výhradou potvrzení správného umístění zkumavky. Rozdrcenou tabletu je třeba podat žaludeční sondou v malém množství vody, poté opláchnout vodou.Po podání rozdrcených potahovaných tablet 15 mg nebo 20 mg přípravku Xarelto by po dávce měla bezprostředně následovat enterální výživa (viz bod 5.2).
04.3 Kontraindikace
Přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Klinicky významné krvácení probíhá.
Zranění nebo stavy, které představují významné riziko velkého krvácení. Mezi ně může patřit nedávná nebo pokračující žaludeční ulcerace, přítomnost maligních novotvarů s vysokým rizikem krvácení, nedávné poranění mozku nebo páteře, mozková, spinální nebo oční operace, nedávné intrakraniální krvácení, známé nebo suspektní varixy jícnu, arteriální-venózní malformace, cévní nebo závažná vaskulární dysfunkce na intraspinální nebo intracerebrální úrovni.
Souběžná léčba jinými antikoagulancii, jako jsou nefrakcionované hepariny, nízkomolekulární hepariny (enoxaparin, dalteparin atd.), Deriváty heparinu (fondaparinux atd.), Perorální antikoagulancia (warfarin, dabigatranetexilát, apixaban atd.), S výjimkou specifický případ změny antikoagulační terapie (viz bod 4.2) nebo pokud jsou nefrakcionované hepariny podávány v dávkách nezbytných k udržení otevřeného centrálního katétru, žilního nebo arteriálního (viz bod 4.5).
Jaterní poruchy spojené s koagulopatií a klinicky významným rizikem krvácení, včetně cirhotických pacientů s Child Pugh B a C (viz bod 5.2).
Těhotenství a kojení (viz bod 4.6).
04.4 Zvláštní upozornění a vhodná opatření pro použití
Po celou dobu léčby se doporučuje sledování podle obvyklé praxe u pacienta na antikoagulační léčbě.
Riziko krvácení
Stejně jako u jiných antikoagulancií by pacienti užívající přípravek Xarelto měli být pečlivě sledováni z hlediska příznaků krvácení. Doporučuje se používat s opatrností v podmínkách zvýšeného rizika krvácení. V případě závažného krvácení musí být podávání přípravku Xarelto přerušeno.
V klinických studiích bylo během dlouhodobé léčby rivaroxabanem častěji ve srovnání s léčbou VKA hlášeno slizniční krvácení (např. Epistaxe, gingivální, gastrointestinální a genitourinární krvácení) a anémie. Pro adekvátní klinický dohled může být důležité v případě potřeby provést laboratorní kontroly hemoglobinu / hematokritu za účelem zjištění skrytého krvácení.
Několik subpopulací pacientů, podrobně popsaných níže, má zvýšené riziko krvácení. Tito pacienti by měli být po zahájení léčby pečlivě sledováni z hlediska známek a příznaků krvácivých komplikací a anémie (viz bod 4.8).
Snížení hemoglobinu nebo krevního tlaku neznámého původu by mělo vést k hledání hemoragického zaměření.
Ačkoli léčba rivaroxabanem nevyžaduje kontinuální sledování expozice, měření hladin rivaroxabanu kalibrovaným kvantitativním testem anti-faktoru Xa může být užitečné ve výjimečných situacích, kdy může být nápomocná znalost expozice rivaroxabanu. Učinit klinické rozhodnutí, jako je předávkování a nouzový chirurgický zákrok (viz. body 5.1 a 5.2).
Porucha funkce ledvin
U pacientů s těžkou poruchou funkce ledvin (clearance kreatininu se plazmatické hladiny rivaroxabanu mohou výrazně zvýšit (v průměru 1,6násobně), což může zvýšit riziko krvácení. Xarelto by měl být používán s opatrností u pacientů s clearance kreatininu mezi 15 a 29 ml / min Nedoporučuje se používat u pacientů s clearance kreatininu
Xarelto by mělo být také používáno s opatrností u pacientů s poruchou funkce ledvin, kteří užívají jiné léčivé přípravky, které zvyšují plazmatické koncentrace rivaroxabanu (viz bod 4.5).
Interakce s jinými léky
Použití přípravku Xarelto se nedoporučuje u pacientů léčených souběžně systémovými azolovými antimykotiky (jako je ketokonazol, itrakonazol, vorikonazol a posakonazol) nebo inhibitory proteázy HIV (např. Ritonavir). Tyto léčivé látky jsou silnými inhibitory CYP3A4 a P-gp a může proto zvýšit plazmatické koncentrace rivaroxabanu v klinicky relevantním rozsahu (v průměru 2,6násobně), což může vést ke zvýšenému riziku krvácení (viz bod 4.5).
Buďte opatrní, pokud jsou pacienti souběžně léčeni léčivými přípravky ovlivňujícími hemostázu, jako jsou nesteroidní protizánětlivé léky (NSAID), kyselina acetylsalicylová a protidestičková léčiva. U pacientů s rizikem peptického vředu lze zvážit vhodnou profylaktickou léčbu (viz bod 4.5).
Jiné rizikové faktory krvácení
Stejně jako u jiných antitrombotik se rivaroxaban nedoporučuje u pacientů se zvýšeným rizikem krvácení, jako například:
• vrozené nebo získané poruchy krvácení
• těžká nekontrolovaná arteriální hypertenze
• jiné gastrointestinální onemocnění bez aktivní ulcerace, které může potenciálně vést ke krvácivým komplikacím (např. Zánětlivé onemocnění střev, ezofagitida, gastritida a gastroezofageální refluxní choroba) vaskulární retinopatie
• bronchiektázie nebo plicní krvácení v anamnéze
Pacienti s ventilovými protézami
Bezpečnost a účinnost přípravku Xarelto nebyla studována u pacientů s protetickými srdečními chlopněmi; proto nejsou k dispozici údaje podporující adekvátní antikoagulační účinek přípravku Xarelto 20 mg (15 mg u pacientů se středně těžkou nebo těžkou poruchou funkce ledvin). V této populaci pacientů. Léčba přípravkem Xarelto se u těchto pacientů nedoporučuje.
Pacienti s hemodynamicky nestabilní PE nebo pacienti vyžadující plicní trombolýzu nebo embolektomii.
Xarelto se nedoporučuje jako alternativa nefrakcionovaného heparinu u pacientů s plicní embolií, kteří jsou hemodynamicky nestabilní nebo kteří mohou podstupovat plicní trombolýzu nebo embolektomii, protože bezpečnost a účinnost přípravku Xarelto nebyla v těchto klinických podmínkách hodnocena.
Spinální / epidurální anestezie nebo punkce
V případě neuraxiální anestézie (spinální / epidurální anestezie) nebo spinální / epidurální punkce jsou pacienti léčení antitrombotickými přípravky k prevenci tromboembolických komplikací ohroženi epidurálním nebo spinálním hematomem, který může způsobit prodlouženou nebo trvalou paralýzu. Toto riziko může být zvýšeno v případě pooperačního použití zavedených epidurálních katetrů nebo kombinovaného použití léčivých přípravků, které mění hemostázu. Riziko se může také zvýšit v případě traumatické nebo opakované epidurální nebo spinální punkce. Pacienti by měli být často sledováni. známky a příznaky neurologických změn (např. necitlivost nebo slabost dolních končetin, dysfunkce střev nebo močového měchýře). V případě neurologického poškození je nutná okamžitá diagnostika a léčba. vztah mezi očekávaným přínosem a rizikem přítomným u pacientů na antikoagulační terapii nebo u pacientů, u nichž je antikoagulační terapie plánována pro antitrombotickou profylaxi. S použitím rivaroxabanu 20 mg v těchto situacích nejsou žádné klinické zkušenosti.
Aby se snížilo potenciální riziko krvácení spojené se souběžným používáním rivaroxabanu a neuraxiální (epidurální / spinální) anestezie nebo páteřní punkce, je třeba zvážit farmakokinetický profil rivaroxabanu. Je vhodnější umístit nebo odstranit epidurální katétr nebo provést punkci . bederní páteře, když je antikoagulační účinek rivaroxabanu odhadován jako nízký. Přesný čas k dosažení dostatečně nízkého antikoagulačního účinku u každého pacienta však není znám.
Pro vyjmutí epidurálního katetru s přihlédnutím k obecným charakteristikám PK by po posledním podání rivaroxabanu mělo uplynout alespoň dvojnásobek poločasu, tj. Alespoň 18 hodin u mladých pacientů a 26 hodin u starších pacientů (viz bod 5.2).
Po vyjmutí katétru musí před podáním další dávky rivaroxabanu uplynout nejméně 6 hodin.
V případě traumatické punkce by mělo být podávání rivaroxabanu odloženo o 24 hodin.
Doporučené dávkování před a po invazivních výkonech a chirurgických zákrocích
Pokud je vyžadován invazivní zákrok nebo chirurgický zákrok, léčba přípravkem Xarelto 20 mg by měla být ukončena, pokud je to možné a na základě klinického úsudku lékaře, nejméně 24 hodin před operací.
Pokud nelze postup odložit, je nutné zvýšené riziko krvácení vyhodnotit ve vztahu k naléhavosti intervence.
Léčba přípravkem Xarelto by měla být obnovena co nejdříve po invazivním zákroku nebo chirurgickém zákroku, jakmile to klinická situace dovolí a bude dosaženo adekvátní hemostázy na základě posouzení lékaře (viz bod 5.2).
Starší populace
Vyšší věk může způsobit zvýšené riziko krvácení (viz bod 5.2).
Informace o pomocných látkách
Xarelto obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
04.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Inhibitory CYP3A4 a P-gp
Souběžné podávání rivaroxabanu a ketokonazolu (400 mg jednou denně) nebo ritonaviru (600 mg dvakrát denně) mělo za následek 2,6 / 2,5násobné zvýšení průměrné AUC rivaroxabanu a 1,7 / 1,6násobek průměrné Cmax rivaroxabanu, s významnou zvýšení farmakodynamických účinků: může to být způsobeno zvýšeným rizikem krvácení. Proto se použití přípravku Xarelto nedoporučuje u pacientů na souběžné systémové léčbě azolovými antimykotiky. jako je ketokonazol, itrakonazol, vorikonazol a posakonazol nebo s HIV proteázou inhibitory. Tyto léčivé látky jsou silnými inhibitory CYP3A4 a P-gp (viz bod 4.4).
Předpokládá se, že účinné látky, které významně inhibují pouze jednu z metabolických cest rivaroxabanu, CYP3A4 nebo P-gp, zvyšují plazmatické koncentrace rivaroxabanu v menší míře. Například klarithromycin (500 mg dvakrát denně), považovaný za silný inhibitor CYP3A4 a slabý až středně silný inhibitor P-gp, vyvolal 1,5násobné zvýšení průměrné AUC rivaroxabanu a zvýšení 1,4násobku Cmax. Toto zvýšení není považováno za klinicky relevantní (pro pacienty s poruchou funkce ledvin: viz bod 4.4).
Erythromycin (500 mg třikrát denně), který mírně inhibuje CYP3A4 a P-gp, vedl k 1,3násobnému zvýšení průměrné AUC a průměrné C rivaroxabanu. Toto zvýšení není považováno za klinicky relevantní.
U subjektů s mírnou poruchou funkce ledvin vyvolal erythromycin (500 mg třikrát denně) průměrné 1,8násobné zvýšení průměrné AUC rivaroxabanu a 1,6násobné zvýšení Cmax ve srovnání s osobami s normální normální funkcí ledvin. U subjektů se středně těžkou poruchou funkce ledvin vyvolal erythromycin průměrné 2,0násobné zvýšení průměrné AUC rivaroxabanu a 1,6násobné zvýšení C ve srovnání s osobami s normální funkcí ledvin. Účinek erythromycinu je aditivní k účinku na renální insuficienci (viz bod 4.4).
Flukonazol (400 mg jednou denně), považovaný za středně silný inhibitor CYP3A4, zvýšil průměrnou AUC rivaroxabanu o 1,4krát a průměrnou Cmax o 1,3krát. Toto zvýšení není považováno za klinicky relevantní. (U pacientů s renální insuficiencí: viz bod 4.4 ).
Vzhledem k omezeným klinickým údajům, které jsou s dronedaronem k dispozici, je třeba se vyvarovat jeho souběžného podávání s rivaroxabanem.
Antikoagulancia
Po souběžném podání enoxaparinu (40 mg jednorázová dávka) a rivaroxabanu (10 mg jednorázová dávka) byl pozorován aditivní účinek na aktivitu anti-faktoru Xa v nepřítomnosti dalších účinků na koagulační testy (PT, aPTT) .Enoxaparin se nezměnil farmakokinetika rivaroxabanu.
Vzhledem ke zvýšenému riziku krvácení je nutná opatrnost v případě souběžné léčby jakýmkoli jiným antikoagulantem (viz body 4.3 a 4.4).
NSAID / protidestičková činidla
Po souběžném podání rivaroxabanu (15 mg) a naproxenu (500 mg) nebylo pozorováno žádné klinicky relevantní prodloužení doby krvácení. Někteří lidé však mohou mít výraznější farmakodynamickou odpověď.
Při současném podávání s rivaroxabanem a 500 mg kyseliny acetylsalicylové nebyly pozorovány žádné klinicky významné farmakokinetické nebo farmakodynamické interakce.
Clopidogrel (nasycovací dávka 300 mg, následovaná udržovací dávkou 75 mg) nevykazoval žádnou farmakokinetickou interakci s rivaroxabanem (15 mg), ale u subpopulace pacientů bylo pozorováno relevantní prodloužení doby krvácení. Nesouvisející se stupněm agregace krevních destiček nebo hladiny P-selektinu nebo receptoru GPIIb / IIIa.
Je třeba opatrnosti, pokud jsou pacienti souběžně léčeni NSAID (včetně kyseliny acetylsalicylové) a protidestičkovými látkami, protože tyto léčivé přípravky obvykle zvyšují riziko krvácení (viz bod 4.4).
Warfarin
Přechod z antagonisty vitaminu K warfarinu (INR mezi 2,0 a 3,0) na rivaroxaban (20 mg) nebo z rivaroxabanu (20 mg) na warfarin (INR mezi 2,0 a 3,0) měl za následek prodloužení protrombinového času / INR (Neoplastin) více než aditivum (lze pozorovat jednotlivé hodnoty INR až 12), zatímco účinky na aPTT, inhibici aktivity faktoru Xa a endogenního trombinového potenciálu (ETP) byly aditiva.
Pokud jsou požadovány farmakodynamické účinky rivaroxabanu během přechodného období, lze použít testy na aktivitu anti-faktoru Xa, PiCT a Heptest, protože nejsou ovlivněny warfarinem. Čtvrtý den po poslední dávce warfarinu., Všechny testy (včetně PT, aPTT, inhibice aktivity faktoru Xa a ETP) odrážejí pouze účinek rivaroxabanu.
Pokud jsou požadovány farmakodynamické účinky warfarinu během přechodného období, lze INR použít při minimální koncentraci (Cvalle) rivaroxabanu (24 hodin po předchozím příjmu rivaroxabanu), protože v té době je tento test rivaroxabanem ovlivněn minimálně.
Mezi warfarinem a rivaroxabanem nebyly pozorovány žádné farmakokinetické interakce.
Induktory CYP3A4
Souběžné podávání rivaroxabanu a silného induktoru CYP3A4 rifampicinu mělo za následek přibližně 50% snížení průměrné AUC rivaroxabanu se souběžným snížením jeho farmakodynamických účinků Souběžné užívání rivaroxabanu a dalších silných induktorů CYP3A4 (např. Fenytoinu, karbamazepinu, fenobarbitalu nebo St Třezalka tečkovaná (Hypericum perforatum)) může snížit plazmatické koncentrace rivaroxabanu. Proto je třeba se vyvarovat podávání silných induktorů CYP3A4, pokud není pacient pečlivě sledován na známky a příznaky trombózy.
Jiné souběžné terapie
Při současném podávání rivaroxabanu a midazolamu (substrát CYP3A4), digoxinu (substrát P-gp), atorvastatinu (substrát CYP3A4 a P-gp) nebo omeprazolu (inhibitor protonové pumpy) nebyly pozorovány žádné klinicky významné farmakokinetické nebo farmakodynamické interakce. Rivaroxaban neinhibuje ani neindukuje žádnou z hlavních izoforem CYP, jako je CYP3A4.
Laboratorní parametry
Koagulační parametry (např. PT, aPTT, HepTest) jsou předvídatelně změněny v důsledku mechanismu účinku rivaroxabanu (viz bod 5.1).
04.6 Těhotenství a kojení
Těhotenství
Bezpečnost a účinnost přípravku Xarelto u těhotných žen nebyla stanovena. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3) .Pro potenciální reprodukční toxicitu, inherentní riziko krvácení a důkaz, že rivaroxaban prochází placentou, je přípravek Xarelto během těhotenství kontraindikován (viz bod 4.3).
Ženy ve fertilním věku by se měly během léčby rivaroxabanem vyhnout otěhotnění.
Čas krmení
Bezpečnost a účinnost přípravku Xarelto u kojících žen nebyla stanovena. Údaje ze zvířat naznačují, že se rivaroxaban vylučuje do lidského mléka. Proto je přípravek Xarelto během laktace kontraindikován (viz bod 4.3). Musí být učiněno rozhodnutí, zda přerušit kojení nebo přerušit / zdržet se léčby.
Plodnost
Nebyly provedeny žádné specifické studie s rivaroxabanem ke stanovení jeho účinků na plodnost u mužů a žen. Ve studii fertility samců a samic na potkanech nebyly pozorovány žádné účinky (viz bod 5.3).
04.7 Účinky na schopnost řídit a obsluhovat stroje
Xarelto má malý vliv na schopnost řídit a obsluhovat stroje.Byly hlášeny nežádoucí účinky jako synkopa (frekvence: méně časté) a závratě (frekvence: časté) (viz bod 4.8) .U pacientů, u kterých se tyto reakce vyskytly nežádoucí účinky, by neměli řídit nebo obsluhovat stroje.
04.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Bezpečnost rivaroxabanu byla stanovena v jedenácti studiích fáze III zahrnujících 32 625 pacientů vystavených rivaroxabanu (viz tabulka 1).
Tabulka 1: Počet studovaných pacientů, maximální denní dávka a délka léčby ve studiích fáze III
* Pacienti vystaveni alespoň jedné dávce rivaroxabanu
Nejčastěji hlášenými nežádoucími účinky u pacientů léčených rivaroxabanem bylo krvácení (viz bod 4.4 a „Popis konkrétních nežádoucích účinků“ níže). Nejčastěji hlášeným krvácením (≥4%) byla epistaxe (5,9%) a krvácení do gastrointestinálního traktu (4,2%).
Celkem byly nežádoucí účinky související s léčbou pozorovány u přibližně 67% pacientů vystavených alespoň jedné dávce rivaroxabanu. Přibližně 22% pacientů mělo nežádoucí účinky, které vyšetřovatelé považovali za související s léčbou. U pacientů léčených 10 mg přípravku Xarelto a podstupujících chirurgickou náhradu kyčle nebo kolena au pacientů hospitalizovaných ze zdravotních důvodů došlo ke krvácení u 6,8% respektive 12,6% pacientů a anémie u 5,9% respektive 2,1% pacientů. . Byli hlášeni pacienti léčení 15 mg přípravku Xarelto dvakrát denně a následně 20 mg jednou denně k léčbě DVT nebo PE nebo 20 mg jednou denně k prevenci recidivy DVT a PE. Krvácení se vyskytlo přibližně u 27,8% pacientů a anémie došlo přibližně u 2,2% pacientů. U pacientů léčených k prevenci cévní mozkové příhody a systémové embolie bylo hlášeno krvácení jakéhokoli typu nebo rozsahu. 28 na 100 pacientoroků a anémii s frekvencí 2,5 na 100 pacientoroků. pacienti léčení k prevenci kardiovaskulární smrti a infarktu myokardu u pacientů po akutním koronárním syndromu (ACS), krvácení na každé úrovni závažnosti, byli hlášeni s frekvencí 22 na 100 pacientoroků. Anemie byla hlášena s frekvencí 1,4 na 100 pacientoroků.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky pozorované u přípravku Xarelto jsou uvedeny v tabulce 2 níže, seřazené podle orgánových systémů (podle MedDRA) a podle frekvence.
Frekvence jsou definovány následovně:
velmi časté (≥ 1/10)
časté (≥ 1/100,
méně časté (≥ 1/1 000,
vzácné (≥ 1/10 000,
velmi vzácné (
není známo (frekvenci nelze z dostupných údajů určit).
Tabulka 2: Všechny nežádoucí účinky související s léčbou hlášené u pacientů ve studiích fáze III
Pozorováno při prevenci žilní tromboembolie (VTE) u dospělých pacientů podstupujících elektivní operaci náhrady kyčle nebo kolena
B pozorované při léčbě DVT a PE a při prevenci relapsů jako velmi časté u žen
C pozorováno jako neobvyklé v prevenci aterotrombotických příhod u pacientů po AKS (po perkutánní koronární intervenci)
Popis konkrétních nežádoucích účinků
Vzhledem k jeho farmakologickému mechanismu účinku může být užívání přípravku Xarelto spojeno se zvýšeným rizikem skrytého nebo zjevného krvácení v jakékoli tkáni nebo orgánu, což může vést k posthemoragické anémii. Příznaky, příznaky a závažnost (včetně smrtelných následků) se liší podle místa a stupně nebo rozsahu krvácení a / nebo anémie (viz bod 4.9 Řízení krvácení). V klinických studiích bylo během dlouhodobé léčby rivaroxabanem častěji ve srovnání s léčbou VKA hlášeno slizniční krvácení (např. Epistaxe, gingivální, gastrointestinální a genitourinární krvácení) a anémie. Pro adekvátní klinický dohled může být důležité v případě potřeby provést laboratorní kontroly hemoglobinu / hematokritu za účelem zjištění skrytého krvácení. Riziko krvácení může být zvýšeno u určitých kategorií pacientů, např. u pacientů s těžkou nekontrolovanou arteriální hypertenzí a / nebo podstupujících souběžnou léčbu s účinky na hemostázu (viz hemoragické riziko v bodě 4.4). Menstruace může mít delší intenzitu a / nebo trvání. Krvácející komplikace se mohou projevit jako slabost, astenie, bledost, závratě, bolest hlavy nebo otok neznámého původu, dušnost a šok neznámého původu.V některých případech byly v důsledku anémie pozorovány příznaky srdeční ischemie, jako je bolest na hrudi nebo angina pectoris.
U přípravku Xarelto byly hlášeny známé komplikace závažného krvácení, jako je kompartment syndrom a poškození ledvin v důsledku hypoperfuze. Při hodnocení stavu pacientů na antikoagulační léčbě by proto měla být zvážena možnost krvácení.
Poznámky post-marketing
Následující nežádoucí účinky byly hlášeny po uvedení přípravku na trh v časové souvislosti s používáním přípravku Xarelto. Četnost těchto nežádoucích účinků hlášených po uvedení přípravku na trh nelze odhadnout.
Poruchy imunitního systému: Angioedém a alergický edém (Ve sloučených studiích fáze III byly tyto příhody méně časté (≥ 1/1 000,
Poruchy jater a žlučových cest: Cholestáza, hepatitida (včetně hepatocelulárního poškození) (Ve sloučených studiích fáze III byly tyto příhody vzácné (≥ 1/10 000,
Poruchy krve a lymfatického systému: Trombocytopenie (ve sloučených studiích fáze III byly tyto příhody méně časté (≥ 1/1 000,
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky, které se vyskytnou po registraci léčivého přípravku, je důležité, protože umožňuje průběžné sledování poměru přínosu a rizika léčivého přípravku. Zdravotničtí pracovníci jsou požádáni, aby hlásili jakékoli podezření na nežádoucí účinky prostřednictvím italské agentury pro léčivé přípravky. , webové stránky: http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Předávkování
Byly hlášeny vzácné případy předávkování až 600 mg bez krvácivých komplikací nebo jiných nežádoucích účinků. Vzhledem k omezené absorpci se očekává stropní účinek bez dalšího zvýšení průměrné plazmatické expozice při supraterapeutických dávkách 50 mg rivaroxabanu nebo vyšších.
Není k dispozici žádné specifické antidotum, které by mohlo antagonizovat farmakodynamické účinky rivaroxabanu.
V případě předávkování rivaroxabanem lze zvážit použití aktivního uhlí ke snížení absorpce.
Řízení krvácení
Pokud se u pacienta léčeného rivaroxabanem objeví komplikace krvácení, následné podání rivaroxabanu by mělo být odloženo nebo léčba podle potřeby přerušena. Rivaroxaban má poločas přibližně 5 až 13 hodin (viz bod 5.2) Léčba pacienta by měla být individualizována podle závažnosti a lokalizace krvácení. Podle potřeby lze provést vhodnou symptomatickou léčbu, jako je mechanická komprese (například v případě závažné epistaxe), chirurgická hemostáza s postupy kontroly krvácení, obnovení tekutin a hemodynamická podpora, podávání krevních produktů (koncentráty erytrocytů nebo čerstvé zmrazené plazmy, v závislosti na přidružené anémii nebo koagulopatii) nebo krevních destičkách.
Pokud nelze krvácení kontrolovat popsanými opatřeními, lze zvážit podání specifického prokoagulačního činidla pro zvrácení antikoagulačního účinku, jako je koncentrát protrombinového komplexu (PCC), koncentrát aktivovaného protrombinového komplexu (APCC).) Nebo rekombinantního faktoru VIIa (r -FVIIa).
K dnešnímu dni jsou však velmi omezené klinické zkušenosti s používáním těchto přípravků u subjektů léčených rivaroxabanem. Doporučení je také založeno na omezených preklinických údajích. Je třeba zvážit opakované podávání rekombinantního faktoru VIIa a upravit jeho dávkování na základě zlepšení krvácení. V případě velkého krvácení by měl být podle místní dostupnosti konzultován odborník na koagulaci (viz bod 5.1).)
Neočekává se, že by protamin sulfát a vitamín K ovlivňovaly antikoagulační aktivitu rivaroxabanu. U subjektů léčených rivaroxabanem jsou omezené zkušenosti s kyselinou tranexamovou, zatímco s kyselinou aminokapronovou a aprotininem nejsou žádné zkušenosti. Neexistuje žádný vědecký důvod pro možný prospěch nebo zkušenost se systémovým hemostatem desmopresinem u subjektů léčených rivaroxabanem. Vzhledem k vysoké vazbě na plazmatické bílkoviny je nepravděpodobné, že by rivaroxaban byl dialyzovatelný.
05.0 FARMAKOLOGICKÉ VLASTNOSTI
05.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Přímý inhibitor faktoru Xa, ATC kód: B01AF01
Mechanismus účinku
Rivaroxaban je přímý a vysoce selektivní inhibitor faktoru Xa s orální biologickou dostupností. Inhibice faktoru Xa narušuje vnitřní i vnější cesty koagulační kaskády a inhibuje jak tvorbu trombinu, tak vývoj trombu Rivaroxaban neinhibuje trombin (aktivovaný faktor II) a nebyl prokázán žádný účinek na krevní destičky.
Farmakodynamické účinky
U lidí byla pozorována na dávce závislá inhibice aktivity faktoru Xa. Protrombinový čas (PT) je při testování s Neoplastinem ovlivňován rivaroxabanem způsobem závislým na dávce, s úzkou korelací s plazmatickými koncentracemi (r rovné 0,98). výsledky jsou získány s jinými činidly. PT musí být vyjádřen v sekundách, protože INR (mezinárodní normalizovaný poměr) je kalibrován a validován pouze pro kumariny a nelze jej použít pro jiná antikoagulancia.
U pacientů léčených rivaroxabanem pro DVT, PE a prevenci relapsu bylo zahrnuto 5/95 percentilů pro PT (Neoplastin) 2 - 4 hodiny po užití tablet (tj. Když je účinek největší). Mezi 17 a 32 s pro 15 mg rivaroxaban dvakrát denně a mezi 15 a 30 s pro 20 mg rivaroxabanu jednou denně. Když je účinek minimální (8 - 16 hodin po užití tablety), percentily 5/95 pro 15 mg dvakrát denně se pohybovaly od 14 do 24 s, zatímco po dobu 20 mg jednou denně (18 až 30 hodin po užití tablety) se pohybovalo od 13 do 20 s.
U pacientů s nevalvulární fibrilací síní léčených rivaroxabanem k prevenci cévní mozkové příhody a systémové embolie se 5/95 percentilů pro PT (Neoplastin) 1 - 4 hodiny po užití tablety (tj. V době maxima účinku) pohybovalo od 14 do 40 s u pacientů léčených 20 mg jednou denně a od 10 do 50 s u pacientů se středně těžkou poruchou funkce ledvin léčených 15 mg jednou denně. Když je účinek minimální (16 - 36 hodin po užití tablety), 5/95 percentilů pro 20 mg jednou denně se pohybovalo od 12 do 26 s a u pacientů se středně těžkou poruchou funkce ledvin léčených 15 mg jednou denně. Časy denně byly mezi 12 a 26 s.
V klinicko -farmakologické studii zkoumající potenciál antagonizovat farmakodynamické účinky rivaroxabanu u zdravých dospělých subjektů (n = 22) byly účinky jednotlivých dávek (50 IU / kg) dvou různých typů PCC, jednoho PCC na 3 faktory (faktory II, IX a X) a 4-faktorový PCC (faktory II, VII, IX a X). 3-faktorový PCC snížil průměrné hodnoty PT s Neoplastinem přibližně o 1,0 sekundu do 30 minut ve srovnání se snížením přibližně o 3,5 sekundy pozorovaným u 4-faktorového PCC. Naproti tomu 3-faktorový PCC měl větší a rychlejší celkový účinek antagonizace změn v tvorbě endogenního trombinu než 4-faktorový PCC (viz bod 4.9). HepTest se zvýšil v rozsahu závislém na dávce; nedoporučují se však pro hodnocení farmakodynamických účinků rivaroxabanu.
V klinické praxi není během léčby rivaroxabanem nutné monitorovat koagulační parametry. Pokud je to však klinicky indikováno, mohou být plazmatické hladiny rivaroxabanu měřeny příslušně kalibrovaným testem anti-faktoru Xa (viz bod 5.2).
Klinická účinnost a bezpečnost
Prevence cévní mozkové příhody a systémové embolie u pacientů s nevalvulární fibrilací síní
Klinický program přípravku Xarelto byl vyvinut s cílem demonstrovat jeho účinnost v prevenci mrtvice a systémové embolie u pacientů s nevalvulární fibrilací síní.
V pivotní dvojitě zaslepené studii ROCKET AF bylo 14 264 pacientů přiřazeno k přípravku Xarelto 20 mg jednou denně (15 mg jednou denně u pacientů s clearance kreatininu 30 - 49 ml / min) nebo k warfarinu titrovanému na cílovou hodnotu INR 2,5 ( terapeutický rozsah 2,0 až 3,0). Medián doby léčby byl 19 měsíců a maximální celková doba léčby byla 41 měsíců.
34,9% pacientů bylo léčeno kyselinou acetylsalicylovou a 11,4% bylo léčeno antiarytmiky třídy III, včetně amiodaronu.
Xarelto nebylo horší než warfarin pro primární složený cílový parametr cévní mozkové příhody a systémové embolie bez CNS. V populaci podle protokolu při léčbě byla u 188 pacientů léčených rivaroxabanem (1,71% ročně) a v 241 pacientů užívajících warfarin (2,16% ročně) (HR 0,79; 95% CI, 0,66-0,96; p
U pacientů léčených warfarinem byly hodnoty INR v terapeutickém rozmezí (2,0 až 3,0) v průměru 55% času (medián, 58%; mezikvartilové rozmezí, 43 až 71). Účinek rivaroxabanu se nelišil v závislosti na střední úrovni TTR (Time in Target INR Range 2,0 až 3,0) u stejně velkých kvartilů (p = 0,74 na interakci). na warfarin bylo 0,74 (95% CI, 0,49 až 1,12).
Míra výskytu pro klíčový bezpečnostní parametr (klinicky relevantní závažné a nevýznamné krvácivé příhody) byla podobná ve dvou léčebných skupinách (viz tabulka 4).
Tabulka 3: Výsledky účinnosti ze studie fáze III ROCKET AF
Tabulka 4: Výsledky bezpečnosti ze studie ROCKET AF fáze III
Pacienti podstupující kardioverzi
Byla provedena prospektivní, randomizovaná, otevřená, multicentrická, zaslepená endpointová (X-VERT) průzkumná studie u 1504 pacientů (nových nebo již na perorální antikoagulační terapii) s nevalvulární fibrilací síní, kteří byli naprogramováni na kardioverzi. bylo porovnat rivaroxaban s dávkou upravenou AVK (randomizace 2: 1) pro prevenci kardiovaskulárních příhod. Použitými strategiemi byla kardioverze vedená TEE (1-5 dní před léčbou) nebo konvenční kardioverze (nejméně tři týdny před léčba).Primární výsledek účinnosti (všechny typy cévních mozkových příhod, přechodný ischemický záchvat, necentrální systémová embolie, srdeční infarkt a kardiovaskulární smrt) se vyskytl u 5 (0,5%) pacientů ve skupině s rivaroxabanem (n = 978) a u 5 (1,0%) pacienti ve skupině AVK (n = 492; RR 0,50; 95%CI 0,15-1,73; modifikovaná populace ITT). Hlavní bezpečnostní výsledek (velké krvácení) se vyskytl u 6 (0,6%) a 4 (0,8%) pacientů v rivaroxabanu skupina (n = 988), respektive skupina AVK (n = 499), (RR 0,76; 95% CI 0,21-2, 67; populace bezpečnosti) Tato průzkumná studie prokázala srovnatelný profil účinnosti a bezpečnosti mezi skupinami léčenými rivaroxabanem a AVK v nastavení kardioverze.
Léčba DVT, PE a prevence recidivy DVT a PE
Klinický program přípravku Xarelto byl vyvinut s cílem demonstrovat jeho účinnost při počáteční a pokračující léčbě akutní DVT a PE a při prevenci recidivy DVT a PE.
Ve třech randomizovaných kontrolovaných klinických studiích fáze III (Einstein DVT, Einstein PE a Einstein Extension) bylo studováno více než 9400 pacientů a byla rovněž provedena předdefinovaná souhrnná analýza studií Einstein DVT a Einstein PE. Maximální celková doba léčby ve všech studiích byla 21 měsíců.
Ve studii Einstein DVT bylo studováno 3 449 pacientů s akutní DVT pro léčbu DVT a prevenci recidivy DVT a PE (pacienti se symptomatickou PE byli ze studie vyloučeni). Délka léčby byla v závislosti na klinickém hodnocení zkoušejícího 3, 6 nebo 12 měsíců.
Po dobu prvních 3 týdnů akutní léčby DVT byl podáván Rivaroxaban 15 mg dvakrát denně. Poté byl podán Rivaroxaban 20 mg jednou denně.
Ve studii Einstein PE bylo studováno 4 832 pacientů s akutním PE pro léčbu PE a prevenci recidivy DVT a PE. Délka léčby byla 3,6 nebo 12 měsíců, na základě posouzení lékařem.
Pro počáteční léčbu akutní PE byl podáván rivaroxaban 15 mg dvakrát denně po dobu tří týdnů, poté rivaroxaban 20 mg jednou denně.
Jak ve studii Einstein DVT, tak ve studii, srovnávací režim sestával z enoxaparinu podávaného po dobu alespoň 5 dnů v kombinaci s antagonisty vitaminu K, dokud nebylo dosaženo PT / INR v terapeutickém rozmezí (≥ 2,0). Léčba pokračuje dávkou antagonisty vitaminu K titrovaného k udržení hodnot PT / INR v terapeutickém rozmezí mezi 2,0 a 3,0.
Ve studii Einstein Extension bylo studováno 1 197 pacientů s DVT nebo PE pro prevenci recidivy DVT a PE. Na základě klinického hodnocení zkoušejícího byla doba léčby prodloužena o 6 nebo 12 měsíců u pacientů, kteří absolvovali 6 až 12 měsíců léčbu žilní tromboembolismu. Přípravek Xarelto 20 mg jednou denně byl srovnáván s placebem.
Ve všech studiích fáze III byly použity stejné předdefinované primární a sekundární cílové parametry účinnosti. Primárním cílovým parametrem účinnosti byl relaps symptomatické VTE, definovaný jako kombinace relapsu DVT a fatální nebo nefatální PE. Sekundární cílový parametr účinnosti byl definován jako kombinace relapsu DVT, nefatálního PE a mortality ze všech příčin.
Ve studii Einstein DVT (viz tabulka 5) byl rivaroxaban v primárním cílovém parametru účinnosti nižší než enoxaparin / VKA (p
Míra výskytu primárních (klinicky relevantních závažných nebo nevýznamných krvácivých příhod) a sekundárních (závažné krvácivé příhody) cílové parametry bezpečnosti byly ve dvou léčebných skupinách podobné.
b) Enoxaparin po dobu nejméně 5 dnů, souběžně a následně AVK * p
Ve studii Einstein PE (viz tabulka 6) byl rivaroxaban v primárním cílovém parametru účinnosti nižší než enoxaparin / VKA (p = 0,0026 (test na noninferioritu); poměr rizik: 1,123 (0,749-1,684)) výchozí čistý klinický přínos (primární cílový parametr účinnosti plus závažné krvácivé příhody) byl hlášen s poměrem rizika 0,849 ((95% CI: 0,633 - 1,139), nominální hodnota p = 0,275). INR byly v terapeutickém rozmezí v průměru 63% doba průměrné doby léčby 215 dní, respektive 57%, 62%a 65%času ve skupinách, jejichž předpokládaná doba léčby byla 3, 6 a 12 měsíců. Ve skupině s enoxaparinem / AVK nebylo jasné vztah mezi průměrnou úrovní TTR centra (Time in Target INR Range mezi 2,0 a 3,0) v tercilech stejné velikosti a výskytu relabujícího VTE (p = 0,082 na interakci) .V rámci nejvyššího tercilu na základě Ve středu byl poměr rizika rivaroxabanu k warfarinu 0,642 (95% CI, 0,277 - 1,484).
Míra incidence primárního cílového parametru bezpečnosti (klinicky relevantní závažné nebo nevýznamné krvácivé příhody) byla o něco nižší ve skupině s rivaroxabanem (10,3% (249/2412)) než ve skupině enoxaparin/VKA (11,4% (274/2405) Výskyt sekundárního cílového parametru bezpečnosti (závažné krvácivé příhody) byl nižší ve skupině s rivaroxabanem (1,1% (26/2412)) než ve skupině s enoxaparinem/AVK (2,2% (52/2405)) s poměrem rizika 0,493 (95% CI: 0,308 - 0,789).
b) Enoxaparin po dobu nejméně 5 dnů, souběžně a následně AVK
Byla provedena předem definovaná souhrnná analýza koncových bodů studií Einstein DVT a PE (viz tabulka 7).
b) Enoxaparin po dobu nejméně 5 dnů, souběžně a následně AVK
Výchozí čistý klinický přínos (primární cílový parametr účinnosti plus závažné krvácivé příhody) v souhrnné analýze byl hlášen s poměrem rizika 0,771 ((95% CI: 0,614 - 0,967), nominální hodnota p = 0,0244).
Ve studii Einstein Extension (viz tabulka 8) byl rivaroxaban v primárních a sekundárních cílových parametrech účinnosti lepší než placebo. U primárního cílového parametru bezpečnosti (závažné krvácivé příhody) byla u pacientů léčených rivaroxabanem v dávce 20 mg jednou denně ve srovnání s placebem pozorována číselně, ale nikoli významně vyšší incidence. míry byly pozorovány u pacientů léčených rivaroxabanem 20 mg jednou denně ve srovnání s placebem.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Xarelto u jedné nebo více podskupin pediatrické populace v léčbě tromboembolických příhod. Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Xarelto u všech podskupin pediatrické populace v prevenci tromboembolických příhod (informace o použití u dětí viz bod 4.2).
05.2 Farmakokinetické vlastnosti
Vstřebávání
Rivaroxaban se rychle vstřebává a vrcholové koncentrace (Cmax) se dosahují 2 - 4 hodiny po užití tablety.
Perorální absorpce rivaroxabanu je téměř úplná a biologická dostupnost při perorálním podání 2,5 mg a 10 mg tablety je vysoká (80 - 100%), bez ohledu na půst nebo příjem potravy. Příjem s jídlem neovlivňuje AUC ani Cmax rivaroxabanu při dávce 2,5 mg a 10 mg.
Kvůli snížené absorpci byla u 20 mg tablety stanovena orální biologická dostupnost 66% za podmínek nalačno. Při užívání tablet Xarelto 20 mg s jídlem bylo pozorováno 39% zvýšení průměrné AUC ve srovnání s užitím tablety nalačno; to naznačuje téměř úplnou absorpci a vysokou orální biologickou dostupnost. Tablety Xarelto 15 mg a 20 mg se užívají s jídlem (viz bod 4.2).
Farmakokinetika rivaroxabanu je zhruba lineární až do přibližně 15 mg jednou denně nalačno. Při příjmu potravy je farmakokinetika úměrná dávce u tablet Xarelto 10 mg, 15 mg a 20 mg. Při vyšších dávkách je absorpce omezena rozpuštěním, což má za následek snížení biologické dostupnosti a rychlosti absorpce. Jak se dávka zvyšuje.
Variabilita ve farmakokinetice rivaroxabanu je střední, s interindividuální variabilitou (CV%) v rozmezí od 30%do 40%.
Absorpce rivaroxabanu závisí na místě uvolňování v gastrointestinálním traktu. Bylo zaznamenáno 29% a 56% snížení AUC a Cmax ve srovnání s tabletou, když se granule rivaroxabanu uvolňují do proximálního tenkého střeva. Expozice se dále snižuje, když je rivaroxaban uvolňován do distálního tenkého střeva nebo vzestupného tračníku. Proto je třeba se vyvarovat podávání rivaroxabanu distálně od žaludku, protože může být snížena absorpce rivaroxabanu a tím i expozice.
Biologická dostupnost (AUC a Cmax) byla srovnatelná pro 20 mg rivaroxabanu podávaného perorálně jako drcené tablety smíchané s jablečným pyré nebo resuspendované ve vodě a podávané sondou následované tekutým jídlem, ve srovnání s celou tabletou. S ohledem na předvídatelný a na dávce závislý farmakokinetický profil rivaroxabanu budou výsledky biologické dostupnosti získané v této studii pravděpodobně platit i pro nižší dávky rivaroxabanu.
Rozdělení
U lidí je vazba na plazmatické bílkoviny vysoká a dosahuje přibližně 92% -95%. Hlavní složkou vazby je sérový albumin. Distribuční objem je mírný s Vss přibližně 50 litrů.
Biotransformace a eliminace
Přibližně 2/3 podané dávky rivaroxabanu podléhají metabolické degradaci; jedna polovina je pak vyloučena ledvinami a druhá polovina fekální cestou. Zbývající 1/3 podané dávky se vylučuje přímo ledvinami jako nezměněná účinná látka močí, zejména aktivní renální sekrecí.
Rivaroxaban je metabolizován prostřednictvím CYP3A4, CYP2J2 a mechanismy nezávislými na CYP. Oxidační degradace morfolinonové skupiny a hydrolýza amidových vazeb jsou hlavními místy biotransformace. Na základě získaných údajů in vitroRivaroxaban je substrátem transportních proteinů P-gp (P-glykoprotein) a Bcrp (protein rezistence na rakovinu prsu).
Nezměněný rivaroxaban je hlavní sloučeninou přítomnou v lidské plazmě, ve které nejsou detekovány žádné důležité nebo aktivní cirkulující metabolity. Se systémovou clearance přibližně 10 l / h lze rivaroxaban popsat jako látku s nízkou clearance. Po intravenózním podání dávky 1 mg je eliminační poločas přibližně 4,5 hodiny. Po perorálním podání je eliminace omezena rychlostí absorpce. K eliminaci rivaroxabanu z plazmy dochází s terminálním poločasem 5 - 9 hodin u mladých subjektů a 11 - 13 hodin u starších osob.
Zvláštní populace
Sex
Mezi pacienty mužského a ženského pohlaví nebyly žádné klinicky významné rozdíly ve farmakokinetice a farmakodynamice.
Starší populace
U starších pacientů byly pozorovány vyšší plazmatické koncentrace než u mladých pacientů s průměrnými hodnotami AUC přibližně 1,5krát vyššími, a to především kvůli (zjevnému) snížení celkové a renální clearance. Úprava dávky není nutná.
Váhové kategorie
Extrémy tělesné hmotnosti (120 kg) měly pouze „snížený“ vliv na plazmatické koncentrace rivaroxabanu (méně než 25%). Úprava dávky není nutná.
Interetnické rozdíly
Pokud jde o farmakokinetiku a farmakodynamiku rivaroxabanu, nebyly mezi kavkazskými, afroamerickými, hispánskými, japonskými nebo čínskými pacienty pozorovány žádné klinicky relevantní interetnické rozdíly.
Porucha funkce jater
Pouze malé změny ve farmakokinetice rivaroxabanu (průměr 1,2násobného zvýšení AUC rivaroxabanu) byly pozorovány u cirhotických pacientů s mírným poškozením jater (klasifikováno jako Child Pugh A), téměř srovnatelné se zdravými kontrolní skupinou. Cirhotičtí pacienti se středně těžkým poškozením jater ( klasifikován jako Child Pugh B), průměrná AUC rivaroxabanu byla významně zvýšena 2,3krát ve srovnání se zdravými dobrovolníky. Nevázaná AUC byla zvýšena 2,6krát. Tito pacienti měli také sníženou renální eliminaci rivaroxabanu, podobně jako pacienti se středně těžkou poruchou funkce ledvin. U pacientů s těžkou poruchou funkce jater nejsou k dispozici žádné údaje.
Inhibice aktivity faktoru Xa byla zvýšena 2,6krát u pacientů se středně těžkou poruchou funkce jater ve srovnání se zdravými dobrovolníky; Prodloužení PT bylo také zvýšeno 2,1krát. Pacienti se středně těžkou poruchou funkce jater byli citlivější na rivaroxaban, což vedlo ke zvýšení sklonu korelační linie PK / PD mezi koncentrací a PT.
Xarelto je kontraindikován u pacientů s jaterním onemocněním spojeným s koagulopatií a klinicky relevantním rizikem krvácení, včetně cirhotických pacientů s Child Pugh B a C (viz bod 4.3).
Porucha funkce ledvin
Na základě stanovení clearance kreatininu došlo ke zvýšení expozice rivaroxabanu v souvislosti se sníženou funkcí ledvin.U subjektů s mírnou (clearance kreatininu 50 - 80 ml / min), středně těžkou (clearance kreatininu 30) poruchou funkce ledvin - 49 ml / min) a závažné (clearance kreatininu 15 - 29 ml / min), plazmatické koncentrace rivaroxabanu (AUC) byly zvýšeny 1,4, 1,5 a 1,6násobně. odpovídající farmakodynamické účinky byly výraznější. U subjektů s lehkou, středně těžkou a těžkou poruchou funkce ledvin byla celková Inhibice aktivity faktoru Xa se zvýšila 1,5krát, 1,9krát a 2,0násobně ve srovnání se zdravými dobrovolníky; PT se podobně zvýšil 1,3krát, 2,2krát a 2,4násobně. U pacientů s clearance kreatininu nejsou k dispozici žádné údaje
Vzhledem k vysoké vazbě na plazmatické bílkoviny se neočekává, že by rivaroxaban byl dialyzovatelný.
Použití u pacientů s clearance kreatininu se nedoporučuje
Farmakokinetické údaje u pacientů
U pacientů léčených rivaroxabanem pro akutní hlubokou žilní trombózu (DVT), kteří dostávali 20 mg jednou denně, byla geometrická průměrná koncentrace (predikční rozmezí 90%) po 2-4 hodinách a přibližně 24 hodin po podání dávky (což přibližně představuje maximální a minimální koncentraci v rozsah příjmu) byl 215 (22 - 535) respektive 32 (6 - 239) mcg / l.
Farmakokinetický / farmakodynamický vztah
Farmakokinetický / farmakodynamický (FC / FD) vztah mezi plazmatickou koncentrací rivaroxabanu a různými koncovými body FD (inhibice faktoru Xa, PT, aPTT, HepTest) byl hodnocen po podání širokého spektra dávek (5 - 30 mg dvakrát denně). Vztah mezi koncentrací rivaroxabanu a aktivitou faktoru Xa je nejlépe popsán modelem Emax. U PT obecně nejlépe popisuje data model lineární regrese. V závislosti na různých použitých činidlech se sklon značně liší. Když byl pro PT použit Neoplastin, základní PT byla asi 13 s a sklon asi 3-4 s / (100 mcg / l). Výsledky analýzy FC / DF ve fázi II a III jsou srovnatelné s údaji získanými od zdravých subjektů.
Pediatrická populace
Bezpečnost a účinnost u dětí a dospívajících do 18 let nebyla ověřena.
05.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje na základě konvenčních studií farmakologické bezpečnosti, toxicity po jedné dávce, fototoxicity, genotoxicity, karcinogenního potenciálu a juvenilní toxicity neodhalily žádné zvláštní riziko pro člověka.
Účinky pozorované ve studiích toxicity po opakovaném podávání byly převážně důsledkem nadměrné farmakodynamické aktivity rivaroxabanu.U potkanů byly při klinicky relevantních úrovních expozice pozorovány zvýšené plazmatické hladiny IgG a IgA.
U potkanů nebyly pozorovány žádné účinky na mužskou ani ženskou plodnost. Studie na zvířatech prokázaly reprodukční toxicitu související s farmakologickým mechanismem účinku rivaroxabanu (např. Krvácivé komplikace). Při klinicky relevantních plazmatických koncentracích byla pozorována embryofetální toxicita (postimplantační ztráta, zpožděná / progresivní osifikace). Více světlých jaterních skvrn ), zvýšený výskyt běžných malformací a abnormalit placenty V prenatální a postnatální studii na potkanech bylo při dávkách toxických pro matku pozorováno snížení životaschopnosti potomstva.
06.0 FARMACEUTICKÉ INFORMACE
06.1 Pomocné látky
Jádro tabletu
Mikrokrystalická celulóza
Sodná sůl kroskarmelózy
Monohydrát laktózy
Hypromelóza
Laurylsulfát sodný
Stearát hořečnatý
Potahovací film
Macrogol 3350
Hypromelóza
Oxid titaničitý (E 171)
Červený oxid železitý (E 172)
06.2 Neslučitelnost
Irelevantní.
06.3 Doba platnosti
3 roky
06.4 Zvláštní opatření pro skladování
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
06.5 Charakter vnitřního obalu a obsah balení
PP / hliníkové blistry v krabičkách po 14, 28 nebo 98 potahovaných tabletách nebo perforované jednodávkové blistry v krabičkách po 10 x 1 potahovaných tabletách, 100 x 1 potahovaných tabletách nebo vícečetném balení obsahujícím 100 potahovaných tablet film (10 balení po 10 x 1).
Na trhu nemusí být všechny velikosti balení.
06.6 Návod k použití a zacházení
Žádné zvláštní pokyny k likvidaci.
07.0 DRŽITEL ROZHODNUTÍ O REGISTRACI
Bayer Pharma AG
13342 Berlín
Německo
08.0 REGISTRAČNÍ ČÍSLO
14 potahovaných tablet blistr (PP / alu) EU / 1/08/472/017 038744177 / E
28 potahovaných tablet blistr (PP / alu) EU / 1/08/472/018 038744189 / E
98 potahovaných tablet blistr (PP / alu) EU / 1/08/472/019 038744191 / E
10 x 1 potahovaný tablet blistr (PP / alu) EU / 1/08/472/020 038744203 / E
100 x 1 potahovaný tablet blistr (PP / alu) EU / 1/08/472/021 038744215 / E
10 x 10 x 1 potahovaný tablet blistr (PP / alu) EU / 1/08/472/024 038744241 / E
09.0 DATUM PRVNÍ REGISTRACE NEBO PRODLOUŽENÍ REGISTRACE
Datum první registrace: 30. září 2008
Datum posledního obnovení: 22. května 2013
10.0 DATUM REVIZE TEXTU
05/2015