Účinné látky: esomeprazol
LUCEN 20 mg enterosolventní tablety
LUCEN 40 mg enterosolventní tablety
Lucen příbalové letáky jsou k dispozici pro velikosti balení: - LUCEN 20 mg enterosolventní tablety, LUCEN 40 mg enterosolventní tablety
- LUCEN 10 mg enterosolventní granule pro perorální suspenzi, ve sáčku
- LUCEN 40 mg prášek pro injekční / infuzní roztok
Indikace Proč se používá Lucen? K čemu to je?
LUCEN obsahuje léčivý přípravek zvaný esomeprazol. Patří do skupiny léků nazývaných „inhibitory protonové pumpy“, které působí snížením množství kyseliny produkované žaludkem.
LUCEN se používá k léčbě následujících poruch:
- „Gastroezofageální refluxní choroba“ (GERD). K tomu dochází, když kyselina ze žaludku uniká do jícnu (trubice, která spojuje hrdlo se žaludkem), což způsobuje bolest, zánět a pálení.
- Žaludeční nebo vředy v horní části střeva infikované bakteriemi zvanými „Helicobacter pylori“. Pokud máte tyto stavy, může vám lékař předepsat také antibiotika k léčbě infekce a umožnění hojení vředu.
- Žaludeční vředy způsobené léky nazývanými NSAID (nesteroidní protizánětlivé léky). LUCEN lze také použít k prevenci vzniku žaludečních vředů při užívání NSAID.
- Přebytek žaludeční kyseliny způsobený nádorem ve slinivce břišní (Zollinger-Ellissonův syndrom).
- Prodloužená léčba opětovného krvácení vředů po prevenci intravenózním podáním Lucenu
Kontraindikace Kdy by Lucen neměl být používán
Neužívejte LUCEN:
- jestliže jste alergický (á) na esomeprazol nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě: Další informace).
- jestliže jste alergický (á) na jiné léky inhibující protonovou pumpu (např. pantoprazol, lansoprazol, rabeprazol, omeprazol).
- jestliže užíváte lék obsahující nelfinavir (používaný k léčbě HIV).
Neměli byste užívat přípravek LUCEN, pokud spadá do některého z výše uvedených případů. Pokud máte pochybnosti, poraďte se před užitím přípravku LUCEN se svým lékařem nebo lékárníkem.
Opatření pro použití Co potřebujete vědět, než začnete užívat Lucen
Zvláštní pozornost věnujte přípravku LUCEN
Před užitím přípravku LUCEN se poraďte se svým lékařem nebo lékárníkem, pokud:
- Máte závažné problémy s játry
- Máte závažné problémy s ledvinami.
LUCEN může maskovat příznaky jiných nemocí. Pokud se vám tedy stane něco z následujícího, než začnete užívat přípravek LUCEN nebo během něj, okamžitě to sdělte svému lékaři:
- Bez příčiny hodně hubnete nebo máte potíže s polykáním
- Objevuje se bolest žaludku nebo zažívací potíže
- Začněte zvracet jídlo nebo krev
- Stolice je černá (stolice potřísněné krví).
Pokud vám byl přípravek LUCEN předepsán „podle potřeby“, obraťte se na svého lékaře, pokud příznaky přetrvávají nebo se mění charakteristiky.
Pokud užíváte inhibitor protonové pumpy, jako je LUCEN, zejména déle než jeden rok, můžete mít mírně zvýšené riziko zlomeniny kyčle, zápěstí nebo páteře. Pokud máte osteoporózu nebo užíváte kortikosteroidy (což může zvýšit riziko osteoporóza) poraďte se se svým lékařem
Interakce Které léky nebo potraviny mohou změnit účinek přípravku Lucen
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte nebo jste v nedávné době užíval, a to i o lécích, které jsou dostupné bez lékařského předpisu.
LUCEN může skutečně ovlivnit způsob účinku některých léků a některé léky mohou mít vliv na přípravek LUCEN. LUCEN byste neměl užívat, pokud užíváte lék obsahující nelfinavir (používaný k léčbě HIV).
Informujte svého lékaře nebo lékárníka, pokud užíváte některý z následujících léků:
- Atazanavir (používá se k léčbě HIV)
- Clopidogrel (používá se k prevenci krevních sraženin)
- Ketokonazol, itrakonazol nebo vorikonazol (používané k léčbě infekcí způsobených houbami).
- Erlotinib (používaný k léčbě rakoviny).
- Citalopram, imipramin nebo klomipramin (používané k léčbě deprese).
- Diazepam (používá se k léčbě úzkosti, k uvolnění svalů nebo při epilepsii).
- Fenytoin (používá se při epilepsii) Pokud užíváte fenytoin, váš lékař vás bude muset sledovat při zahájení nebo ukončení léčby přípravkem LUCEN.
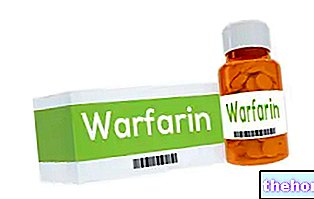
- Léky používané k ředění krve, jako je warfarin. Váš lékař vás může sledovat, když zahájíte nebo ukončíte léčbu přípravkem LUCEN.
- Cilostazol (používá se k léčbě přerušované klaudikace - bolesti nohou při chůzi kvůli nedostatečnému prokrvení).
- Cisaprid (používá se při poruchách trávení a pálení žáhy).
- Digoxin (používá se při srdečních problémech).
- Methotrexát (chemoterapeutický lék používaný ve vysokých dávkách k léčbě rakoviny) - pokud užíváte vysoké dávky methotrexátu, lékař vám může dočasně ukončit léčbu přípravkem Lucen.
- Takrolimus (používá se při transplantacích orgánů)
- Rifampicin (používá se k léčbě tuberkulózy)
- Třezalka tečkovaná (Hypericum perforatum) (používá se k léčbě deprese).
Pokud vám lékař předepsal antibiotika jako amoxicilin a klarithromycin s přípravkem LUCEN k léčbě vředů způsobených infekcí Helicobacter pylori, je velmi důležité, abyste svému lékaři sdělili další léky.
Varování Je důležité vědět, že:
Těhotenství a kojení
Před užitím přípravku LUCEN informujte svého lékaře, pokud jste těhotná nebo chcete otěhotnět. Před užitím jakéhokoli léku se poraďte se svým lékařem nebo lékárníkem. Váš lékař rozhodne, zda můžete během této doby užívat LUCEN.
Není známo, zda přípravek LUCEN přechází do mateřského mléka, proto byste přípravek LUCEN neměl užívat, pokud kojíte.
Užívání přípravku LUCEN s jídlem a pitím
Tablety lze užívat na plný žaludek nebo na prázdný žaludek.
Řízení dopravních prostředků a obsluha strojů
LUCEN pravděpodobně neovlivní vaši schopnost řídit nebo obsluhovat jakékoli nástroje nebo stroje.
Důležité informace o některých složkách přípravku LUCEN
LUCEN enterosolventní tablety obsahují sacharózu, což je druh cukru. Pokud vám lékař řekl, že trpíte „nesnášenlivostí některých cukrů, poraďte se s ním před užitím léku.
Dávkování a způsob použití Jak používat Lucen: Dávkování
Vždy užívejte přípravek LUCEN přesně podle pokynů svého lékaře. Pokud máte pochybnosti, poraďte se se svým lékařem nebo lékárníkem.
- Gastro-rezistentní tablety LUCEN se nedoporučují pro děti do 12 let
- Pokud tento lék užíváte delší dobu, lékař vás bude sledovat (zvláště pokud jej užíváte déle než rok)
- Pokud vám lékař řekl, abyste užíval (a) lék podle potřeby, sdělte to svému lékaři, pokud se vaše příznaky změní.
Užívání léku
- Tablety můžete užít kdykoli během dne.
- Tablety můžete užívat na plný žaludek nebo na prázdný žaludek.
- Tablety spolkněte celé a zapijte vodou. Tablety nežvýkejte ani nedrťte, protože obsahují obalené granule, které chrání léčivo před žaludeční kyselostí, a proto je důležité granule nepoškodit.
Co dělat, pokud máte potíže s polykáním tablet
Pokud máte potíže s polykáním tablet:
- Tablety vložte do sklenice neperlivé vody. Nepoužívejte jiné kapaliny
- Míchejte, dokud se tablety nerozpustí (směs nebude mít jasný vzhled). Pijte okamžitě nebo alespoň do 30 minut. Před pitím je vždy promíchejte
- Abyste se ujistili, že jste užili veškerý lék, důkladně vypláchněte sklenici naplněním do poloviny vodou a nápojem. Pevné částice obsahují lék a neměly by se žvýkat ani drtit.
Pokud absolutně nemůžete polykat, lze tabletu smíchat s trochou vody, vložit do injekční stříkačky a podat hadičkou přímo do žaludku (žaludeční trubice).
Kolik léků vzít
- Váš lékař vám poradí, kolik tablet máte užívat a jak dlouho. Je to funkce vašeho fyzického stavu, věku a stavu jater.
- Obvyklé dávky jsou uvedeny níže.
Léčba pálení žáhy způsobená gastroezofageální refluxní chorobou (GERD):
Dospělí a děti od 12 let:
- Pokud váš lékař zjistí, že je váš jícen mírně poškozený, obvyklá dávka je jedna 40mg enterosolventní tableta LUCEN jednou denně po dobu 4 týdnů. Váš lékař vám může říci, abyste pokračovali v léčbě stejnou dávkou po dobu dalších 4 týdnů, pokud se jícen nezhojí.
- Po zhojení jícnu je obvyklá dávka jedna 20 mg enterosolventní tableta LUCEN jednou denně.
- Pokud není jícen poškozen, je obvyklá dávka jedna enterosolventní tableta LUCEN 20 mg denně. Když jsou vaše příznaky pod kontrolou, lékař vás bude informovat, že v případě potřeby můžete lék užít, maximálně však jednu gastro -odolná tableta. Lucen 20 mg denně.
- Pokud máte závažné problémy s játry, lékař vám podá nižší dávku.
Léčba vředů způsobených infekcí Helicobacter pylori a prevence jejich opětovného výskytu:
- Dospělí od 18 let dále: obvyklá dávka je jedna enterosolventní tableta LUCEN 20 mg dvakrát denně po dobu jednoho týdne.
- Váš lékař vám také řekne, abyste užívali antibiotika zvaná amoxicilin a klarithromycin.
Léčba žaludečních vředů způsobených NSAID (nesteroidní protizánětlivá léčiva):
- Dospělí od 18 let věku: obvyklá dávka je jedna enterosolventní tableta LUCEN 20 mg jednou denně po dobu 4 až 8 týdnů.
Prevence žaludečních vředů, pokud užíváte NSAID (nesteroidní protizánětlivá léčiva):
- Dospělí od 18 let výše: obvyklá dávka je jedna enterosolventní tableta LUCEN 20 mg jednou denně.
Léčba přebytečné žaludeční kyseliny způsobené růstem slinivky břišní (Zollinger-Ellisonův syndrom):
- Dospělí od 18 let dále: obvyklá dávka je jedna 40mg enterosolventní tableta LUCEN dvakrát denně.
- Váš lékař upraví dávku podle vaší potřeby a také rozhodne, jak dlouho v léčbě pokračovat.
Maximální dávka je 80 mg dvakrát denně.
Prodloužená léčba opětovného krvácení vředů po prevenci intravenózním podáním Lucenu:
Obvyklá dávka je jedna 40mg tableta přípravku Lucen jednou denně po dobu 4 týdnů.
Předávkování Co dělat, když jste užil příliš mnoho přípravku Lucen
Jestliže jste užil (a) více LUCENU, než jste měl (a)
Jestliže jste užil (a) více přípravku LUCEN, než Vám předepsal lékař, ihned to sdělte svému lékaři nebo lékárníkovi.
Jestliže jste zapomněl (a) užít LUCEN
- Pokud zapomenete užít dávku přípravku LUCEN, užijte ji, jakmile si vzpomenete. Pokud je téměř čas na další dávku, vynechejte zapomenutou dávku.
- Nezdvojnásobujte následující dávku (dvě dávky současně), abyste nahradil (a) zapomenutou dávku.
Nežádoucí účinky Jaké jsou vedlejší účinky přípravku Lucen
Podobně jako všechny léky, může mít i LUCEN nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud zaznamenáte některý z následujících závažných nežádoucích účinků, přestaňte přípravek LUCEN užívat a okamžitě kontaktujte svého lékaře:
- Náhlé sípání, otok rtů, jazyka a hrdla nebo těla, vyrážka, mdloby nebo potíže s polykáním (závažná alergická reakce).
- Zčervenání kůže s puchýři nebo odlupováním. Silné puchýře a krvácení se mohou objevit také na rtech, očích, ústech, nosu a genitáliích. Může to být „Stevens-Johnsonův syndrom“ nebo „toxická epidermální nekrolýza“.
- Žlutá kůže, tmavá moč a únava mohou být příznaky problémů s játry. Tyto účinky jsou vzácné, postihují méně než 1 z 1000 lidí.
Mezi další nežádoucí účinky patří:
Časté (postihují méně než 1 z 10 lidí):
- Bolest hlavy.
- Účinky na žaludek nebo střeva: průjem, bolest žaludku, zácpa, plynatost.
- Nevolnost nebo zvracení.
Méně časté (postihují méně než 1 ze 100 lidí):
- Otoky nohou a kotníků.
- Zhoršený spánek (nespavost).
- Závratě, mravenčení, ospalost.
- Závrať.
- Suchá ústa
- Změny v krevních testech, které kontrolují fungování jater.
- Kožní vyrážka, kopřivka a svědění.
- Zlomenina kyčle, zápěstí nebo páteře (pokud se Lucen používá ve vysokých dávkách a po delší dobu).
Vzácné (postihují méně než 1 z 1000 lidí):
- Krevní problémy, jako je snížený počet bílých krvinek a krevních destiček. To může způsobit slabost, podlitiny nebo usnadnit infekce.
- Nízké hladiny sodíku v krvi. To může způsobit slabost, zvracení a křeče.
- Pocit rozrušení, zmatenosti nebo deprese.
- Změny chuti.
- Problémy se zrakem, například rozmazané vidění.
- Náhlé sípání nebo dušnost (bronchospasmus).
- Zánět vnitřku úst.
- Infekce zvaná „drozd“, která může postihnout střevo a je způsobena houbou.
- Problémy s játry, včetně žloutenky, která může způsobit žlutou kůži, tmavou moč a únavu.
- Vypadávání vlasů (alopecie).
- Kožní vyrážka na slunci.
- Bolest kloubů (artralgie) nebo bolest svalů (myalgie).
- Celkový pocit nevolnosti a nedostatku síly.
- Zvýšené pocení.
Velmi vzácné (postihují méně než 1 z 10 000 lidí):
- Změny v počtu krvinek, včetně agranulocytózy (nedostatek bílých krvinek).
- Agrese.
- Vidět, cítit nebo slyšet věci, které neexistují (halucinace).
- Závažné problémy s játry vedoucí k selhání jater a zánětu mozku.
- Náhlý nástup závažné vyrážky nebo puchýřů nebo olupování kůže. To může být spojeno s vysokou horečkou a bolestmi kloubů (multiformní erytém, Stevens-Johnsonův syndrom, toxická epidermální nekrolýza).
- Svalová slabost.
- Závažné problémy s ledvinami.
- Zvětšení prsou u mužů.
Není známo (frekvenci nelze z dostupných údajů určit)
- Pokud užíváte přípravek LUCEN déle než tři měsíce, může dojít ke snížení hladiny hořčíku v krvi. Nízká hladina hořčíku se může projevit únavou, nedobrovolnými svalovými stahy, dezorientací, křečemi, závratěmi, zvýšenou srdeční frekvencí. Pokud máte některý z těchto příznaků, okamžitě se poraďte se svým lékařem. Nízké hladiny hořčíku mohou také vést ke snížení hladiny draslíku nebo vápníku v krvi. Váš lékař by měl rozhodnout, zda vám bude pravidelně kontrolovat hladinu hořčíku v krvi.
- Zánět ve střevě (vedoucí k průjmu).
LUCEN může ve velmi vzácných případech postihnout bílé krvinky vedoucí k imunodeficienci. Pokud máte infekci s příznaky, jako je horečka se závažným zhoršením celkového fyzického stavu nebo horečka s příznaky lokální infekce, jako je bolest v krku, v krku nebo v ústech nebo potíže s močením, měli byste co nejdříve navštívit svého lékaře. že nedostatek bílých krvinek (agranulocytóza) lze vyloučit krevním testem. Je pro vás důležité poskytnout informace o lécích, které užíváte. Nebojte se výše uvedeného seznamu možných nežádoucích účinků. Může se stát, že se u vás žádný neobjeví. Pokud některý z nežádoucích účinků začne být závažný, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, sdělte to prosím svému lékaři nebo lékárníkovi.
Expirace a retence
- Uchovávejte mimo dosah a dohled dětí.
- Uchovávejte při teplotě do 30 ° C.
- Uchovávejte v původním obalu (blistru) nebo uchovávejte obal těsně uzavřený (lahvička), aby byl přípravek chráněn před vlhkostí.
- Nepoužívejte tablety po uplynutí doby použitelnosti (EXP), uvedené na krabičce, peněžence nebo blistru. Doba použitelnosti se vztahuje k poslednímu dni v měsíci.
- Léčivé přípravky se nesmí vyhazovat do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Pomůže to chránit životní prostředí.
JINÁ INFORMACE
Co přípravek LUCEN obsahuje
Léčivou látkou je esomeprazol. Enterosolventní tablety LUCEN jsou k dispozici ve 2 silách obsahujících 20 nebo 40 mg esomeprazolu (jako trihydrát hořečnatého).
Dalšími složkami jsou: glycerol monostearát 40-55, hyprolosa, hypromelóza, oxid železitý (červenohnědý, žlutý) (E172, pouze pro 20 mg tablety), stearát hořečnatý, kopolymer kyseliny methakrylové, ethylakrylát (1: 1) disperze při 30 ° C %, mikrokrystalická celulóza, syntetický parafín, makrogoly, polysorbát 80, krospovidon, natrium -stearylfumarát, sacharosové kuličky (sacharóza a kukuřičný škrob), mastek, oxid titaničitý (E171), triethylcitrát.
Popis vzhledu LUCEN a obsahu balení
- LUCEN 20 mg enterosolventní tablety jsou světle růžové s A / EH na jedné straně a 20 mg na druhé.
- LUCEN 40 mg enterosolventní tablety jsou růžové s A / EI na jedné straně a 40 mg na druhé.
- Tablety jsou v blistrech, peněženkách a / nebo lahvích obsahujících
- 20 mg, 40 mg: lahvička 2-5-7-14-15-28-30-56-60-100-140 (28x5) tablet.
- 20 mg, 40 mg: blistr nebo peněženka v blistru s 3-7-7x1-14-15-25x1-28-30-50x1- 56-60-90-98-100x1-140 tabletami.
Na trhu nemusí být všechny velikosti balení
Zdroj příbalové informace: AIFA (Italská agentura pro léčivé přípravky). Obsah zveřejněný v lednu 2016. Přítomné informace nemusí být aktuální.
Chcete-li mít přístup k nejaktuálnější verzi, doporučujeme navštívit webovou stránku AIFA (Italská agentura pro léčivé přípravky). Prohlášení a užitečné informace.
01.0 NÁZEV LÉČIVÉHO PŘÍPRAVKU
LUCENTIS 10 MG / ML ROZTOK PRO INJEKCI
02.0 KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje 10 mg ranibizumabu *. Jedna injekční lahvička obsahuje 2,3 mg ranibizumabu v 0,23 ml roztoku.
* Ranibizumab je fragment humanizované monoklonální protilátky produkovaný v buňkách Escherichia coli technologií rekombinantní DNA.
Úplný seznam pomocných látek viz bod 6.1.
03.0 LÉKOVÁ FORMA
Injekční roztok
Čirý, bezbarvý až světle žlutý vodný roztok.
04.0 KLINICKÉ INFORMACE
04.1 Terapeutické indikace
Lucentis je indikován u dospělých k:
• Léčba neovaskulární (vlhké) makulární degenerace související s věkem (AMD)
• Léčba poškození zraku způsobeného diabetickým makulárním edémem (DME)
• Léčba poškození zraku způsobeného „makulárním edémem sekundárním po okluzi sítnicové žíly (větev RVO nebo centrální RVO)
• Léčba poškození zraku způsobeného choroidální neovaskularizací (CNV) sekundárně k patologické myopii (PM)
04.2 Dávkování a způsob podání
Lucentis by měl být podáván kvalifikovaným oftalmologem se zkušenostmi s intravitreálními injekcemi.
Dávkování pro léčbu vlhké AMD
Doporučená dávka Lucentisu je 0,5 mg podávaná měsíčně jako jedna intravitreální injekce. To odpovídá injektovanému objemu 0,05 ml.
Léčba se podává měsíčně a pokračuje, dokud není dosaženo maximální zrakové ostrosti, tj. Zraková ostrost pacienta je stabilní po dobu tří po sobě jdoucích měsíčních kontrol prováděných během léčby ranibizumabem.
Proto by měla být zraková ostrost pacientů monitorována každý měsíc.
Léčba by měla být obnovena, pokud sledování indikuje pokles zrakové ostrosti v důsledku vlhké AMD. Poté by měly být aplikovány měsíční injekce, dokud nebude znovu dosaženo stabilní zrakové ostrosti po dobu tří po sobě jdoucích měsíčních kontrol (to znamená minimálně dvě injekce). Interval mezi dvěma dávkami by neměl být kratší než jeden měsíc.
Dávkování pro léčbu poškození zraku způsobeného DME nebo makulárním edémem sekundárním k RVO
Doporučená dávka Lucentisu je 0,5 mg podávaná měsíčně jako jedna intravitreální injekce. To odpovídá injektovanému objemu 0,05 ml.
Léčba se podává měsíčně a pokračuje, dokud není dosaženo maximální zrakové ostrosti, tj. Zraková ostrost pacienta je stabilní po dobu tří po sobě jdoucích měsíčních kontrol prováděných během léčby ranibizumabem. Pokud během prvních tří injekcí nedojde ke zlepšení zrakové ostrosti, pokračování léčby se nedoporučuje.
Proto by měla být zraková ostrost pacientů monitorována každý měsíc.
Léčba by měla být obnovena, pokud monitorování indikuje sníženou zrakovou ostrost v důsledku DME nebo makulárního edému sekundárního k RVO. Měsíční injekce by pak měly být podávány, dokud nebude znovu dosaženo stabilní zrakové ostrosti po dobu tří měsíčních kontrol. Po sobě jdoucích (to zahrnuje minimálně dvě injekce). Interval mezi dvěma dávkami by neměl být kratší než jeden měsíc.
Lucentis a laserová fotokoagulace v DME a makulární edém sekundární k BRVO
Existují určité zkušenosti s podáváním Lucentisu souběžně s laserovou fotokoagulací (viz bod 5.1). Pokud je Lucentis podáván ve stejný den, měl by být podán nejméně 30 minut po laserové fotokoagulaci. Lucentis lze podávat pacientům, kteří již dříve podstoupili laserovou fotokoagulaci.
Dávkování pro léčbu poškození zraku způsobeného CNV sekundárně k PM
Léčba by měla být zahájena jednou injekcí.
Pokud sledování odhalí známky aktivity onemocnění, jako je snížená zraková ostrost a / nebo známky poranění, doporučuje se další léčba.
Monitorování onemocnění může zahrnovat klinické vyšetření, optickou koherentní tomografii (OCT) nebo fluoresceinovou angiografii (FA).
Zatímco někteří pacienti mohou potřebovat pouze jednu nebo dvě injekce během prvního roku léčby, někteří mohou potřebovat častější léčbu (viz bod 5.1). První dva měsíce a nejméně každé tři měsíce během prvního roku léčby se proto doporučuje sledovat každý měsíc. Po prvním roce může frekvenci sledování stanovit lékař.
Interval mezi dvěma dávkami by neměl být kratší než jeden měsíc.
Lucentis a fotodynamická terapie Visudynem v CNV sekundárně k PM
Nejsou žádné zkušenosti s podáváním Lucentisu v kombinaci s Visudynem.
Zvláštní populace
Jaterní nedostatečnost
Lucentis nebyl studován u pacientů s jaterní insuficiencí. Pro tuto polaci však nejsou zapotřebí žádné zvláštní úvahy.
Selhání ledvin
U pacientů s renální insuficiencí není nutná úprava dávky (viz bod 5.2).
Senioři
U starších osob není nutná úprava dávky. U pacientů s DME nad 75 let jsou „omezené“ zkušenosti.
Pediatrická populace
Bezpečnost a účinnost přípravku Lucentis u dětí a dospívajících mladších 18 let nebyla stanovena. Nejsou k dispozici žádné údaje.
Způsob podání
Jednorázové injekční lahvičky pouze pro intravitreální použití.
Před podáním by měl být Lucentis vizuálně zkontrolován na přítomnost částic a změnu barvy.
Postup pro injekci musí být proveden za aseptických podmínek, které zahrnují dezinfekci rukou jako u jakéhokoli chirurgického zákroku, sterilní rukavice, sterilní roušku a sterilní blefarostat (nebo ekvivalent) a možnost provedení sterilní paracentézy (v případě potřeby pacientova anamnéza reakcí přecitlivělosti by měla být před intravitreálním postupem pečlivě vyhodnocena (viz bod 4.4). Podle klinické praxe by měla být před injekcí podána adekvátní anestezie a širokospektrální lokální antimikrobiální látka k dezinfekci periokulárního, očního a očního povrchu.
Informace o přípravě přípravku Lucentis viz bod 6.6.
Vložte injekční jehlu 3,5-4,0 mm za limbus, do sklivcové komory, vyhněte se horizontálnímu poledníku a nasměrujte jehlu do středu oční bulvy. Vstříkněte injekční objem 0,05 ml; pro další injekce změňte místo skléry.
04.3 Kontraindikace
Přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Pacienti se současnými nebo suspektními očními nebo periokulárními infekcemi.
Pacienti s pokračujícím závažným nitroočním zánětem.
04.4 Zvláštní upozornění a vhodná opatření pro použití
Reakce související s intravitreální injekcí
Intravitreální injekce, včetně injekcí s Lucentisem, byly spojeny s endoftalmitidou, nitroočním zánětem, rhegmatogenním odchlípením sítnice, rupturou sítnice a iatrogenní traumatickou kataraktou (viz bod 4.8). K podávání přípravku Lucentis by měly být vždy použity vhodné aseptické injekční techniky. Kromě toho by měli být pacienti v týdnu po injekci sledováni, aby bylo možné rychlé léčení v případě infekce. Pacienti by měli být poučeni o tom, jak bezodkladně hlásit jakékoli příznaky naznačující endoftalmitidu nebo některou z výše uvedených příhod.
Zvýšení nitroočního tlaku
Během 60 minut po injekci přípravku Lucentis bylo pozorováno přechodné zvýšení nitroočního tlaku (IOP). Bylo také pozorováno prodloužené zvýšení nitroočního tlaku (viz bod 4.8). Nitrooční tlak a perfúzi hlavy zrakového nervu je třeba monitorovat a vhodně léčit.
Oboustranná léčba
Omezené údaje o dvoustranném používání přípravku Lucentis (včetně dávkování ve stejný den) nenaznačují zvýšené riziko systémových nežádoucích účinků ve srovnání s jednostrannou léčbou.
Imunogenita
U přípravku Lucentis existuje potenciál pro imunogenicitu. Jelikož existuje možnost zvýšené systémové expozice u subjektů s DME, nelze vyloučit zvýšené riziko vzniku přecitlivělosti v této populaci pacientů.Pacienti by měli být také poučeni o tom, jak hlásit, pokud se nitrooční zánět zhorší, protože by to mohl být klinický příznak způsobený tvorba nitroočních protilátek.
Souběžné použití s jinými anti-VEGF (vaskulární endoteliální růstový faktor)
Lucentis nesmí být podáván současně s jinými léčivými přípravky proti VEGF (systémovými nebo očními).
Přerušení Lucentis
Dávka by neměla být podána a léčba by neměla být obnovena před další plánovanou léčbou v případě:
• pokles nejlépe korigované zrakové ostrosti (BCVA) o ≥ 30 písmen ve srovnání s posledním hodnocením;
• nitrooční tlak ≥30 mmHg;
• retinální zlomenina;
• „subretinální krvácení zasahující do středu fovey, nebo pokud je rozsah krvácení ≥ 50% celkové plochy léze“;
• nitrooční chirurgický zákrok provedený nebo plánovaný během předchozích nebo následujících 28 dnů.
Ruptura sítnicového pigmentového epitelu
Mezi rizikové faktory spojené s nástupem ruptury pigmentového epitelu sítnice po terapii vlhkou AMD s anti-VEGF patří velké a / nebo vysoké odchlípení pigmentového epitelu sítnice. Při zahájení léčby přípravkem Lucentis je třeba postupovat opatrně u pacientů s těmito rizikovými faktory pro prasknutí sítnicového pigmentového epitelu.
Regmatogenní odchlípení sítnice nebo makulární otvory
Léčba by měla být ukončena u jedinců s rhegmatogenním odchlípením sítnice nebo stádiem 3 nebo 4 makulárních děr.
Populace s omezenými údaji
Existují pouze omezené zkušenosti s léčbou subjektů s DME sekundárním diabetem typu I. Lucentis nebyl studován u pacientů, kteří dříve dostali intravitreální injekce, u pacientů s aktivními systémovými infekcemi, proliferativní diabetickou retinopatií nebo u pacientů se souběžnými zdravotními stavy jako je odchlípení sítnice nebo makulární díra Rovněž nejsou žádné zkušenosti s léčbou Lucentisem u diabetických pacientů s HbAlc vyšším než 12% a nekontrolovanou hypertenzí. Při léčbě těchto pacientů by měl lékař vzít v úvahu nedostatek informací.
U pacientů s PM jsou k dispozici omezené údaje o účinku Lucentisu u pacientů dříve léčených neúspěšnou fotodynamickou terapií verteporfinem (vPDT) .Kromě toho byl pozorován konzistentní účinek u subjektů s subfoveálními a juxtafoveálními lézemi, ale nejsou k dispozici dostatečné údaje o účinek Lucentisu u subjektů PM s extrafoveálními lézemi.
Systémové efekty po intravitreálním podání
Po intravitreální injekci inhibitorů VEGF byly hlášeny systémové nežádoucí účinky včetně neokulárního krvácení a arteriálních tromboembolických příhod.
Údaje o bezpečnosti léčby DME, makulárního edému způsobeného RVO a CNV sekundárně po PM u pacientů s anamnézou cévní mozkové příhody nebo přechodných ischemických záchvatů jsou omezené. Při léčbě takových pacientů je nutná zvláštní opatrnost (viz bod 4.8).
Předchozí epizody RVO, ischemické větve a centrálního RVO
Zkušenosti s léčbou pacientů s předchozími epizodami RVO a pacientů s ischemickou větví RVO (BRVO) a centrálním RVO (CRVO) jsou omezené. není doporučeno.
04.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné konvenční studie interakcí.
Kombinované použití fotodynamické terapie (PDT) s verteporfinem a Lucentisem u vlhké AMD a PM viz bod 5.1.
Kombinované použití laserové fotokoagulace a přípravku Lucentis v léčbě DME a BRVO viz body 4.2 a 5.1.
04.6 Těhotenství a kojení
Ženy ve fertilním věku / antikoncepce u žen
Ženy ve fertilním věku by měly během léčby používat účinnou antikoncepci.
Těhotenství
Pro ranibizumab nejsou k dispozici žádné klinické údaje o těhotenství během těhotenství. Studie na opicích cynomolgus neprokázaly žádné přímé ani nepřímé škodlivé účinky na březost nebo embryonální / fetální vývoj (viz bod 5.3). Systémová expozice ranibizumabu je po očním podání nízká, ale vzhledem k mechanismu účinku by měl být ranibizumab považován za potenciálně teratogenní a embryo / fetotoxický. Ranibizumab by proto neměl být používán během těhotenství, pokud očekávaný přínos nepřeváží potenciální riziko pro plod. Ženám, které plánují otěhotnět a které byly léčeny ranibizumabem, se doporučuje počkat alespoň 3 měsíce po poslední dávce ranibizumabu, než počne dítě.
Těhotenství
Není známo, zda se Lucentis vylučuje do lidského mléka. Při používání přípravku Lucentis se nedoporučuje kojit.
Plodnost
Nejsou k dispozici žádné údaje o plodnosti.
04.7 Účinky na schopnost řídit a obsluhovat stroje
Procedura léčby přípravkem Lucentis může vyvolat přechodné poruchy vidění, které mohou ovlivnit schopnost řídit nebo obsluhovat stroje (viz bod 4.8). Pacienti s těmito příznaky by neměli řídit ani obsluhovat stroje, dokud tyto přechodné poruchy zraku neustanou.
04.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Většina nežádoucích účinků hlášených po podání Lucentisu souvisí s postupem intravitreální injekce.
Nejčastěji hlášenými očními nežádoucími reakcemi po injekci Lucentisu jsou: bolest očí, oční hyperémie, zvýšený nitrooční tlak, vitreitida, odloučení sklivce, krvácení do sítnice, poruchy vidění, plováky (sklivcové plováky), spojivkové krvácení, podráždění oka, pocit cizího tělesa v oko, zvýšené slzení, blefaritida, suché oko a svědivé oko.
Nejčastěji hlášenými neokulárními nežádoucími účinky jsou bolest hlavy, nazofaryngitida a artralgie.
Méně často hlášené, ale závažnější nežádoucí účinky zahrnují endoftalmitidu, slepotu, odchlípení sítnice, rupturu sítnice a iatrogenní traumatickou kataraktu (viz bod 4.4).
Pacienti by měli být informováni o příznacích těchto potenciálních nežádoucích účinků a měli by být poučeni, aby informovali svého lékaře, pokud se u nich objeví příznaky, jako je bolest očí nebo zvýšené nepohodlí, zhoršení zarudnutí očí, rozmazané nebo zhoršené vidění, zvýšený počet sklivcových plováků nebo „ zvýšená citlivost na světlo.
Nežádoucí účinky hlášené po podání přípravku Lucentis v klinických studiích jsou shrnuty v následující tabulce.
Tabulka nežádoucích účinků #
Nežádoucí účinky jsou seřazeny podle třídy orgánových systémů a frekvence pomocí následující konvence: velmi časté (≥1 / 10), časté (≥1 / 100,
Infekce a infestace
Velmi časté Nasofaryngitida
běžný Infekce močových cest *
Poruchy krve a lymfatického systému
běžný Anémie
Poruchy imunitního systému
běžný Přecitlivělost
Psychiatrické poruchy
běžný Úzkost
Poruchy nervového systému
Velmi časté Bolest hlavy
Oční poruchy
Velmi časté Vitreitida, odchlípení sklivce, krvácení do sítnice, poruchy vidění, bolest očí, sklivcové plováky, krvácení do spojivky, podráždění očí, pocit cizího tělesa v oku, zvýšené slzení, blefaritida, suché oko, oční hyperémie, svědivé oko.
běžný Retinální degenerace, poruchy sítnice, odchlípení sítnice, odtržení sítnice, odchlípení sítnicového pigmentového epitelu, roztržení sítnicového pigmentového epitelu, zhoršená zraková ostrost, sklivcové krvácení, poruchy sklivce, uveitida, iritida, iridocyklitida, katarakta, subkapsulární katarakta zadní kapsle, punktát keratitida, odření rohovky, reakce přední komory, rozmazané vidění, krvácení v místě vpichu, oční krvácení, zánět spojivek, zánět spojivek
alergický, oční výtok, světelné záblesky, fotofobie, oční diskomfort, edém očních víček, bolest očních víček, hyperémie spojivky.
Méně časté Slepota, endoftalmitida, hypopion, hyfém, keratopatie, synechie duhovky, ložiska rohovky, edém rohovky, strie v rohovce, bolest v místě vpichu, podráždění v místě vpichu, abnormální pocity v oku, podráždění očních víček.
Respirační, hrudní a mediastinální poruchy
běžný Kašel
Gastrointestinální poruchy
běžný Nevolnost
Poruchy kůže a podkožní tkáně
běžný Alergické reakce (vyrážka, kopřivka, svědění, erytém)
Poruchy svalové a kosterní soustavy a pojivové tkáně
Velmi časté Artralgie
Diagnostické testy
Velmi časté Zvýšený nitrooční tlak
# Nežádoucí účinky byly definovány jako nežádoucí účinky (u alespoň 0,5 procentního bodu pacientů), které se vyskytovaly častěji (alespoň 2 procentní body) u pacientů léčených přípravkem Lucentis 0,5 mg ve srovnání s těmi, kteří absolvovali kontrolní léčbu (simulovaná nebo PDT) verteporfin).
* pozorováno pouze v populaci s DME
Nežádoucí účinky související s kategorií léčiv
Ve studiích mokré AMD fáze III byla celková frekvence neokulárního krvácení, což je nežádoucí účinek potenciálně související s inhibitory VEGF (růstový faktor endotelových cév), mírně zvýšena u pacientů léčených ranibizumabem. Neexistuje však konzistentní vzor mezi různými krváceními. V důsledku intravitreálního použití inhibitorů VEGF existuje teoretické riziko arteriálních tromboembolických příhod, včetně mrtvice a infarktu myokardu. V klinických studiích s Lucentisem byl u pacientů s AMD, DME, RVO a PM pozorován nízký výskyt arteriálních tromboembolických příhod a mezi skupinami s ranibizumabem nebyly ve srovnání s kontrolou pozorovány žádné rozdíly.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky, které se vyskytnou po registraci léčivého přípravku, je důležité, protože umožňuje průběžné sledování poměru přínosu a rizika léčivého přípravku. Zdravotničtí pracovníci jsou požádáni, aby hlásili jakékoli podezření na nežádoucí účinky prostřednictvím italské agentury pro léčivé přípravky. , web: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Předávkování
Případy náhodného předávkování byly hlášeny z klinických studií s vlhkou AMD a z údajů po uvedení na trh. Nežádoucími účinky nejčastěji spojenými s těmito případy byly zvýšený nitrooční tlak, přechodná slepota, snížená zraková ostrost, edém rohovky a bolest. Pokud dojde k předávkování, nitrooční tlak by měl být monitorován a léčen podle potřeby lékaře.
05.0 FARMAKOLOGICKÉ VLASTNOSTI
05.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, anti-neovaskulární látky, ATC kód: S01LA04
Ranibizumab je humanizovaný fragment rekombinantní monoklonální protilátky namířený proti lidskému vaskulárnímu endoteliálnímu růstovému faktoru A (VEGF-A). Váže se s vysokou afinitou na izoformy VEGF-A (např. VEGF110, VEGF121 a VEGF165), čímž brání navázání VEGF-A na jeho receptory VEGFR-1 a VEGFR-2. Na jeho receptory vede k proliferaci endoteliálních buněk na neovaskularizace a ke zvýšení vaskulární permeability, o nichž se předpokládá, že přispívají k progresi neovaskulární formy věkem podmíněné makulární degenerace, patologické myopie nebo sníženého vidění způsobeného buď diabetickým makulárním edémem nebo „makulárním edémem sekundárním k RVO.
Léčba vlhké AMD
U vlhké AMD byla bezpečnost a klinická účinnost přípravku Lucentis hodnocena ve třech 24měsíčních randomizovaných, dvojitě zaslepených, falešně nebo aktivně kontrolovaných studiích u pacientů s neovaskulární AMD. Do těchto studií bylo zařazeno celkem 1 323 pacientů (879 léčených a 444 kontrol).
Ve studii FVF2598g (MARINA) dostalo 716 pacientů s minimálně klasickými nebo okultními lézemi choroidální neovaskularizace (CNV) bez klasické složky měsíčně intravitreální injekce Lucentisu 0,3 mg (n = 238) nebo 0,5 mg (n = 240) nebo fingované injekce (n = 238).
Ve studii FVF2587g (ANCHOR) dostalo 423 pacientů s převážně klasickou CNV jednu z následujících léčby: 1) měsíční intravitreální injekce Lucentis 0,3 mg a PDT simulované (n = 140); 2) měsíční intravitreální injekce Lucentis 0,5 mg a PDT simulované (n = 140); nebo 3) intravitreální simulované injekce a PDT s verteporfinem (n = 143). PDT s verteporfinem nebo simulovaným podáním bylo podáno společně s počáteční injekcí přípravku Lucentis a následně každé 3 měsíce, pokud fluorangiografie ukázala perzistenci nebo obnovení vaskulárního prosakování.
Klíčová zjištění jsou shrnuta v tabulkách 1, 2 a na obrázku 1.
Tabulka 1 Výsledky ve 12. a 24. měsíci ve studii FVF2598g (MARINA)
ap
Tabulka 2 Výsledky ve 12. a 24. měsíci ve studii FVF2587g (ANCHOR)
Stůl
Výsledky obou studií ukázaly, že pokračující léčba ranibizumabem by mohla být přínosem také pro pacienty, kteří v prvním roce léčby ztratili ≥15 písmen s nejlépe korigovanou zrakovou ostrostí (BCVA).
Studie FVF3192g (PIER) byla randomizovaná, dvojitě zaslepená, falešně kontrolovaná studie navržená k vyhodnocení bezpečnosti a účinnosti přípravku Lucentis u 184 pacientů se všemi formami neovaskulární AMD. Pacienti dostali intravitreální injekci Lucentis 0,3 mg (n = 60) nebo 0,5 mg (n = 61) nebo simulované injekce (n = 63) jednou měsíčně po dobu 3 po sobě jdoucích dávek, po nichž následuje jedna dávka podávaná jednou za 3 měsíce. Od 14. měsíce studie byli pacienti léčeni simulovanou injekcí přijímáni do léčbu ranibizumabem a od 19. měsíce bylo možné provádět častější ošetření. Pacienti léčení přípravkem Lucentis ve studii PIER absolvovali v průměru 10 ošetření.
Primárním cílovým parametrem účinnosti byla průměrná změna zrakové ostrosti po 12 měsících ve srovnání s výchozím stavem. Po počátečním zvýšení zrakové ostrosti (po měsíční dávce) se zraková ostrost pacientů v průměru snížila se čtvrtletním dávkováním, vrátila se na výchozí hodnotu ve 12. měsíci a tento účinek byl u většiny léčených pacientů zachován. S ranibizumabem (82%) při Měsíc 24. Údaje od omezeného počtu subjektů, kteří byli převedeni na léčbu ranibizumabem po více než jednom roce fingované léčby, naznačovaly, že časné zahájení léčby může být spojeno s lepším zachováním „zrakové ostrosti“.
Ve studiích MARINA a ANCHOR bylo zlepšení zrakové ostrosti pozorované u přípravku Lucentis 0,5 mg po 12 měsících doprovázeno přínosy hlášenými pacientem, měřeno skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Rozdíly mezi Lucentis 0,5 mg a dvě kontrolní skupiny byly hodnoceny s hodnotami p v rozmezí od 0,009 do
Účinnost přípravku Lucentis při léčbě vlhké AMD byla potvrzena v postmarketingových studiích AMD.Údaje ze dvou studií (MONT BLANC, BPD952A2308 a DENALI, BPD952A2309) neprokázaly další účinky kombinovaného podávání verteporfinu (Visudyne PDT) a Lucentis ve srovnání samotnému Lucentisovi.
Léčba poškození zraku v důsledku DME
Bezpečnost a účinnost přípravku Lucentis byla hodnocena ve dvou randomizovaných, dvojitě zaslepených, falešně kontrolovaných nebo aktivních 12měsíčních studiích u pacientů se zhoršeným viděním v důsledku diabetického makulárního edému. Celkem bylo zařazeno těchto studií. 496 pacientů (336 aktivních a 160 kontrol), většina měla diabetes typu II, 28 léčených pacientů mělo diabetes typu I.
Ve fázi II studie D2201 (RESOLVE) bylo 151 pacientů léčeno ranibizumabem (6 mg / ml, n = 51, 10 mg / ml, n = 51) nebo simulovaným (n = 49) jednou „intravitreální injekcí za měsíc. dokud nebyla splněna předdefinovaná kritéria. Počáteční dávku ranibizumabu (0,3 mg nebo 0,5 mg) bylo možné kdykoli během studie po první injekci zdvojnásobit. Laserová fotokoagulace byla povolena jako záchranná léčba od 3. měsíce do obou léčebných ramen. Studie měl dvě části: průzkumnou část (prvních 42 pacientů navštívilo v 6. měsíci) a potvrzovací část (zbývajících 109 pacientů navštívilo ve 12. měsíci).
Klíčová zjištění z potvrzující části studie (2/3 pacientů) jsou shrnuta v tabulce 3.
Tabulka 3 Výsledky ve 12. měsíci ve studii D2201 (RESOLVE) (celková populace studie)
ap
Ve studii fáze D2301 fáze III (RESTORE) bylo randomizováno 345 pacientů se zrakovým postižením v důsledku makulárního edému, kteří dostali buď „intravitreální injekci 0,5 mg ranibizumabu v monoterapii a laserovou simulovanou fotokoagulaci (n = 116), nebo kombinaci 0,5 mg ranibizumabu a laserová fotokoagulace (n = 118) nebo falešná injekce a laserová fotokoagulace (n = 111). Léčba ranibizumabem byla zahájena měsíčními intravitreálními injekcemi a pokračovala, dokud zraková ostrost nezůstala stabilní po dobu alespoň tří po sobě následujících měsíčních kontrol. Léčba byla obnovena, když bylo pozorováno snížení BCVA v důsledku progrese DME. Stejný den byla na počátku podána laserová fotokoagulace, nejméně 30 minut před injekcí ranibizumabu a poté podle potřeby na základě kritérií ETDRS.
Klíčová zjištění jsou shrnuta v tabulce 4 a na obrázku 2.
Tabulka 4 Výsledky ve 12. měsíci ve studii D2301 (OBNOVIT)
ap
Účinek byl konzistentní ve většině podskupin.
Zlepšení zrakové ostrosti ve 12. měsíci pozorované u přípravku Lucentis 0,5 mg bylo doprovázeno pacientem hlášenými přínosy hlavních funkcí souvisejících se zrakem, měřeno skóre National Eye Institute Visual Function Questionnaire (VFQ-25). Žádné rozdíly způsobené léčbou nemohly být Rozdíl mezi Lucentis 0,5 mg a kontrolní skupinou byl pro kompozitní skóre VFQ-25 hodnocen s hodnotou p 0,0137 (ranibizumab mono) a 0,0041 (ranibizumab + laser).
V obou studiích bylo vizuální zlepšení doprovázeno kontinuálním snižováním makulárního edému měřeného jako tloušťka centrální sítnice (CRT).
Léčba poškození zraku způsobeného makulárním edémem sekundárním k RVO
Klinická bezpečnost a účinnost přípravku Lucentis u pacientů se zrakovým postižením způsobeným sekundárním makulárním edémem byla hodnocena v randomizovaných, dvojitě zaslepených, kontrolovaných studiích: BRAVO a CRUISE, do kterých byli zařazeni pacienti s BRVO (n = 397) a CRVO (n = 392 V obou studiích dostali pacienti buď intravitreální nebo simulované injekce ranibizumabu 0,3 mg nebo 0,5 mg. Po 6 měsících byli pacienti ve fiktivní kontrolní skupině přesunuti do skupiny ranibizumab 0,5 mg. Ve studii BRAVO byla laserová fotokoagulace jako záchranná léčba byl povolen do všech zbraní od 3. měsíce.
Klíčová zjištění ze studií BRAVO a CRUISE jsou uvedena v tabulkách 5 a 6
Tabulka 5 Výsledky v 6. a 12. měsíci (BRAVO)
ap
Tabulka 6 Výsledky v 6. a 12. měsíci (CRUISE)
ap
V obou studiích bylo vizuální zlepšení doprovázeno kontinuálním a významným snížením makulárního edému měřeného z hlediska tloušťky centrální sítnice.
U pacientů s BRVO (studie BRAVO a prodloužení studie HORIZON): Po 2 letech měli pacienti, kteří byli v prvních 6 měsících léčeni simulovanými injekcemi a následně přešli na léčbu ranibizumabem, zisk AV (& symp; 15 písmen) srovnatelný s pacientů, kteří byli léčeni ranibizumabem od zahájení studie (& 16 písmen). Počet pacientů, kteří dokončili 2 roky, byl však omezený a ve studii HORIZON byly naplánovány pouze čtvrtletní návštěvy. existuje dostatek důkazů k uzavření doporučení kdy má být léčba ranibizumabem zahájena u pacientů s BRVO.
U pacientů s CRVO (studie CRUISE a rozšíření studie HORIZON): Po 2 letech pacienti, kteří byli v prvních 6 měsících léčeni falešnými injekcemi a následně převedeni na léčbu ranibizumabem, nevykazovali žádné zisky v AV (& 6 písmena) ve srovnání s těmi pacientů, kteří byli od zahájení studie léčeni ranibizumabem (& symp; 12 písmen).
Zlepšení zrakové ostrosti pozorované při léčbě ranibizumabem v měsících 6 a 12 bylo doprovázeno přínosy hlášenými pacienty, jak bylo měřeno podskupinou dotazníků Národních očních ústavů pro vizuální funkce (NEI VFQ-25) pro blízké a vzdálené aktivity Rozdíl mezi Lucentis 0,5 mg a kontrolní skupina byla mezi hodnotami p mezi 0,02 a 0,0002.
Léčba poškození zraku v důsledku CNV sekundárně k PM
Bezpečnost a klinická účinnost přípravku Lucentis u pacientů se zrakovým postižením způsobeným CNV u PM byla ověřena na základě 12měsíčních údajů z pivotní randomizované, dvojitě zaslepené, kontrolované studie F2301 (RADIANCE). Tato studie měla za cíl vyhodnotit dva různé dávkovací režimy ranibizumabu 0,5 mg podávaného intravitreální injekcí versus verteporfin PDT (vPDT, fotodynamická terapie Visudyne). 277 pacientů bylo randomizováno do jednoho z následujících ramen:
• Skupina I (ranibizumab 0,5 mg, léčebný režim určený kritérii „stability“ definovanými jako žádná změna v BCVA ve srovnání s hodnoceními z předchozích dvou měsíců).
• Skupina II (ranibizumab 0,5 mg, léčebný režim určený kritérii „aktivity onemocnění“ definovanými jako poškození zraku způsobené intra- nebo subretinální tekutinou nebo aktivní únik způsobený lézemi CNV, jak dokazuje OCT a / nebo AF).
• Skupina III (pacienti léčení vPDT - s možností léčby ranibizumabem od 3. měsíce).
Během 12 měsíců studie dostali pacienti průměrně 4,6 injekce (rozmezí 1–11) ve skupině I a 3,5 injekce (rozmezí 1–12) ve skupině II. U pacientů patřících do skupiny II, která odráží doporučené dávkování (viz bod 4.2), podstoupilo 50,9% pacientů léčbu 1 až 2 injekcemi, 34,5% 3 až 5 injekcí a 14,7% podalo 6 až 12 injekcí během 12měsíční studie. . 62,9% pacientů skupiny II nevyžadovalo žádné injekce během druhých 6 měsíců studie.
Klíčová zjištění z RADIANCE jsou shrnuta v tabulce 7 a na obrázku 5.
Tabulka 7 Výsledky ve 3. a 12. měsíci (RADIANCE)
ap
b Srovnávací kontrola do 3. měsíce. Pacienti randomizovaní k podávání vPDT byli způsobilí pro léčbu ranibizumabem ve 3. měsíci (ve skupině III dostalo 38 pacientů ranibizumab ve 3. měsíci)
Zlepšení vidění bylo doprovázeno snížením tloušťky centrální sítnice.
Ve srovnání se skupinou léčenou vPDT vykazovali pacienti ve skupinách léčených ranibizumabem přínos (hodnota p
Pediatrická populace
Bezpečnost a účinnost ranibizumabu u dětí nebyla dosud stanovena.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Lucentis u všech podskupin pediatrické populace pro neovaskulární AMD, poškození zraku v důsledku DME, poškození zraku v důsledku sekundárního makulárního edému na RVO a poškození zraku v důsledku CNV sekundární PM (informace o použití u dětí viz bod 4.2).
05.2 Farmakokinetické vlastnosti
Po měsíčním intravitreálním podávání přípravku Lucentis pacientům s neovaskulární AMD byly sérové koncentrace ranibizumabu obecně nízké, přičemž vrcholové hladiny (Cmax) byly obecně nižší než koncentrace ranibizumabu potřebné k inhibici biologické aktivity VEGF o 50% (11- 27 ng / ml, hodnoceno v testu in vitro buněčná proliferace). Cmax byla úměrná dávce v celém rozsahu dávek 0,05 až 1,0 mg / oko.U omezeného počtu pacientů s DME zjištěné sérové koncentrace naznačují, že nelze vyloučit o něco vyšší systémovou expozici než u pacientů s neovaskulární AMD. Sérové koncentrace ranibizumabu u pacientů s RVO byly podobné nebo mírně vyšší než koncentrace pozorované u pacientů s neovaskulární AMD.
Na základě populační farmakokinetické analýzy a sérové clearance ranibizumabu u neovaskulárních pacientů s AMD léčených dávkou 0,5 mg je průměrný poločas eliminace sklivce ranibizumabu přibližně 9 dní. V době měsíčního intravitreálního podávání Lucentisu 0,5 mg / oko se očekává, že sérová C ranibizumabu, dosažená přibližně 1 den po podání, se bude obecně pohybovat mezi 0,79 a 2,90 ng / ml, přičemž se očekává, že Cmin obecně kolísá mezi 0,07 a 0,49 ng / ml. Odhaduje se, že sérové koncentrace ranibizumabu jsou přibližně 90 000krát nižší než koncentrace sklivce.
Pacienti s renální insuficiencí: Nebyly provedeny žádné konvenční studie zkoumající farmakokinetiku Lucentisu u pacientů s renální insuficiencí.Ve „farmakokinetické analýze v populaci neovaskulárních pacientů s AMD mělo 68% (136 z 200) pacientů„ renální insuficienci (46,5% mírné [50–80 ml / min], 20% střední [30–50 ml / min] a 15% závažných [systémová clearance byla o něco nižší, ale to nebylo klinicky významné.
Pacienti s jaterní insuficiencí: Nebyly provedeny žádné konvenční studie zkoumající farmakokinetiku Lucentisu u pacientů s jaterní insuficiencí.
05.3 Předklinické údaje vztahující se k bezpečnosti
Oboustranné intravitreální podávání ranibizumabu opicím makaků v dávkách mezi 0,25 mg / oko a 2,0 mg / oko jednou za 2 týdny po dobu až 26 týdnů mělo za následek oční efekty závislé na dávce.
Intraokulárně došlo k nárůstu vzplanutí a buněk závislých na dávce v přední komoře, což vrcholilo 2 dny po injekci. Závažnost zánětlivé odpovědi obecně klesá s následnými injekcemi nebo během období zotavení. V zadním segmentu došlo k buněčné infiltraci a sklivcovým plavákům, který také míval závislost na dávce a obecně přetrvával až do konce léčebného období.V 26týdenní studii se závažnost zánětu sklivce zvyšovala s počtem injekcí. Po období zotavení však byla pozorována reverzibilita. Povaha a trvání zánětu zadního segmentu svědčí o imunitně zprostředkované protilátkové odpovědi, která může být klinicky irelevantní. U některých zvířat byla po relativně dlouhém období intenzivního zánětu pozorována tvorba katarakty, což naznačuje, že změny čočky byly sekundární po závažném zánětu. Po podání bylo pozorováno přechodné zvýšení nitroočního tlaku bez ohledu na dávku po intravitreálních injekcích.
Mikroskopické oční změny souvisely se zánětem a neindikovaly degenerativní procesy. Zánětlivé granulomatózní změny byly zaznamenány v optickém disku některých očí. Tyto změny zadního segmentu se během období zotavení zmenšily a v některých případech odezněly.
Po intravitreálním podání nebyly zaznamenány žádné známky systémové toxicity. Sérum a sklivcové protilátky proti ranibizumabu byly nalezeny v podskupině léčených zvířat.
Nejsou k dispozici žádné údaje o karcinogenitě nebo mutagenitě.
U gravidních opic nezpůsobila intravitreální injekce ranibizumabu vedoucí k maximální systémové expozici 0,9-7násobku nejhorší klinické expozice vývojovou toxicitu nebo teratogenitu a neměla žádný vliv na hmotnost nebo strukturu těla. Placenta, i když ranibizumab by měl být považován za potenciálně teratogenní a embryo / fetotoxický na základě svého farmakologického účinku.
Absence zprostředkovaných účinků ranibizumabu na vývoj embrya / plodu věrohodně souvisí hlavně s neschopností fragmentu Fab překročit placentu. Byl však popsán případ s vysokými hladinami ranibizumabu v séru matky a přítomností ranibizumabu ve fetálním séru, což naznačuje, že protilátka proti ranibizumabu působila jako protein (obsahující oblast FC), který transportuje ranibizumab, čímž se snižuje jeho eliminace z mateřského séra a umožnění jeho přenosu do placenty. Protože testy na vývoj embrya / plodu byly provedeny na zdravých březích zvířatech a některé nemoci (jako je diabetes) mohou modifikovat placentární permeabilitu pro fragment Fab, musí být studie interpretována opatrně.
06.0 FARMACEUTICKÉ INFORMACE
06.1 Pomocné látky
dihydrát α, α-trehalózy
Histidin hydrochlorid, monohydrát
Histidin
Polysorbát 20
Voda na injekci
06.2 Neslučitelnost
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
06.3 Doba platnosti
3 roky
06.4 Zvláštní opatření pro skladování
Uchovávejte v chladničce (2 ° C - 8 ° C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
06.5 Charakter vnitřního obalu a obsah balení
0,23 ml sterilního roztoku v injekční lahvičce (sklo typu I) se zátkou (chlorbutylová pryž), 1 tupou filtrační jehlou (18 G x 1½ ", 1,2 mm x 40 mm, 5 mcm), 1 injekční jehlou (30 G x ½", 0,3 mm x 13 mm) a 1 injekční stříkačku (polypropylen) (1 ml). Balení obsahuje 1 lahvičku.
06.6 Návod k použití a zacházení
Injekční lahvička, injekční jehla, filtrační jehla a stříkačka jsou pouze na jedno použití. Opakované použití může způsobit infekci nebo jiné onemocnění / zranění. Všechny komponenty jsou sterilní. Nesmí být použity žádné součásti s obalem, které vykazují známky poškození nebo nedovolené manipulace. Sterilitu nelze zaručit, pokud těsnění obalu součásti není neporušené.
Při přípravě Lucentisu na intravitreální injekci postupujte podle následujících pokynů:
1. Před odběrem dezinfikujte vnější stranu gumové zátky lahvičky.
2. Asepticky nasaďte 5mcm filtrační jehlu (18G x 1½ ", 1,2 mm x 40 mm, součást dodávky) na 1 ml stříkačku (součást dodávky). Zasuňte tupou filtrační jehlu do středu víčka, dokud se nedotkne dna lahvičky.
3. Natáhněte veškerou tekutinu z lahvičky tak, že ji budete držet ve vzpřímené poloze, mírně nakloněné, aby se usnadnilo úplné natažení.
4. Ujistěte se, že píst stříkačky je při vyprazdňování injekční lahvičky zatažen dostatečně daleko, aby došlo k úplnému vyprázdnění jehly filtru.
5. Ponechte jehlu filtru tupou v lahvičce a vyjměte z ní stříkačku. Po vyjmutí obsahu lahvičky zlikvidujte jehlu filtru a nepoužívejte ji k intravitreální injekci.
6. Nasaďte injekční jehlu (30 G x ½ ", 0,3 mm x 13 mm, součást dodávky) bezpečně a asepticky na injekční stříkačku.
7. Opatrně sejměte víčko z injekční jehly, aniž byste injekční jehlu ze stříkačky odpojili.
Poznámka: Při odstraňování víčka držte žlutou základnu injekční jehly.
8. Opatrně vytlačte vzduch ze stříkačky a upravte dávku na 0,05 ml vyznačenou na stříkačce Stříkačka je připravena k injekci.
Poznámka: Nečistěte injekční jehlu, netahejte píst zpět.
Po injekci nezakrývejte jehlu ani ji neodpojujte od stříkačky. Zlikvidujte použitou injekční stříkačku spolu s jehlou ve vhodném kontejneru nebo v souladu s místními požadavky.
07.0 DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
Wimblehurst Road
Horsham
West Sussex, RH12 5AB
Spojené království
08.0 REGISTRAČNÍ ČÍSLO
EU/1/06/374/001
037608027
09.0 DATUM PRVNÍ REGISTRACE NEBO PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. ledna 2007
Datum posledního obnovení: 24. ledna 2012
10.0 DATUM REVIZE TEXTU
05/2014


















.jpg)
