Aktivní složky: Infliximab
Remicade 100 mg prášek pro koncentrát pro infuzní roztok
Proč se používá přípravek Remicade? K čemu to je?
Remicade obsahuje léčivou látku infliximab. Infliximab je protein lidského a zvířecího (z myší) původu.
Remicade patří do skupiny léků nazývaných „blokátory TNF“. Používá se u dospělých k léčbě následujících zánětlivých onemocnění:
- Revmatoidní artritida
- Psoriatická artritida
- Ankylozující spondylitida (Bechtěrevova choroba)
- Lupénka
Remicade se také používá u dospělých a dětí ve věku 6 let a starších k:
- Crohnova nemoc
- Ulcerózní kolitida.
Remicade působí tak, že blokuje působení proteinu zvaného „tumor necrosis factor alfa“ (TNFα). Tento protein se podílí na zánětlivých procesech těla a jeho zablokováním je možné snížit zánět v těle.
Revmatoidní artritida
Revmatoidní artritida je zánětlivé onemocnění kloubů. Pokud máte revmatoidní artritidu, budete zpočátku léčeni jinými léky. Pokud na tyto léky nereagujete adekvátně, budete léčeni přípravkem Remicade v kombinaci s jiným lékem nazývaným methotrexát pro:
- Snižte příznaky a symptomy onemocnění,
- Zpomalte postup poškození kloubů,
- Zlepšit fyzickou funkci.
Psoriatická artritida
Psoriatická artritida je zánětlivé onemocnění kloubů, obvykle doprovázené lupénkou. Pokud máte psoriatickou artritidu, budete nejprve léčeni jinými léky. Pokud na tyto léky nereagujete adekvátně, budete léčeni přípravkem Remicade:
- Snižte příznaky a symptomy onemocnění,
- Zpomalte postup poškození kloubů,
- Zlepšit fyzickou funkci.
Ankylozující spondylitida (Bechtěrevova choroba)
Ankylozující spondylitida je zánětlivé onemocnění páteře. Pokud máte ankylozující spondylitidu, budete nejprve léčeni jinými léky. Pokud na tyto léky nereagujete dostatečně, budete léčeni přípravkem Remicade:
- Snižte příznaky a symptomy onemocnění,
- Zlepšit fyzickou funkci.
Lupénka
Psoriáza je zánětlivé kožní onemocnění. Pokud máte středně těžkou až těžkou ložiskovou psoriázu, budete nejprve léčeni jinými léky nebo jinými způsoby léčby, jako je fototerapie. Pokud na tyto léky nebo léčbu nereagujete adekvátně, budete léčeni přípravkem Remicade ke snížení projevů a příznaků vašeho onemocnění.
Ulcerózní kolitida
Ulcerózní kolitida je zánětlivé onemocnění střev. Pokud máte ulcerózní kolitidu, budete nejprve léčeni jinými léky. Pokud na tyto léky nereagujete adekvátně, dostanete k léčbě onemocnění Remicade.
Crohnova nemoc
Crohnova choroba je zánětlivé onemocnění střev. Pokud máte Crohnovu chorobu, budete nejprve léčeni jinými léky. Pokud na tyto léky nereagujete adekvátně, budete léčeni přípravkem Remicade k: • léčbě aktivní Crohnovy choroby • snížení počtu abnormálních otvorů (píštělí) mezi střevem a kůží, pro které se jiné léky nebo chirurgické zákroky ukázaly jako nedostatečné.
Kontraindikace Kdy by přípravek Remicade neměl být používán
Remicade byste neměli dostat, pokud:
- jste alergický na infliximab (léčivá látka v přípravku Remicade) nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jste alergický (přecitlivělý) na myší bílkoviny
- máte tuberkulózu (TBC) nebo „jinou závažnou infekci, jako je zápal plic nebo sepse“
- mít „srdeční selhání, které je středně závažné nebo závažné.
Neužívejte Remicade, pokud se na vás vztahuje některá z výše uvedených podmínek. Pokud si nejste jisti, poraďte se před podáním přípravku Remicade se svým lékařem
Opatření pro použití Co potřebujete vědět před užitím přípravku Remicade
Před podáním přípravku Remicade se poraďte se svým lékařem, pokud máte:
Dříve obdržel Remicade
- Informujte svého lékaře, pokud jste v minulosti byli léčeni přípravkem Remicade a zda léčbu přípravkem Remicade znovu zahájíte.
Pokud jste přestali užívat přípravek Remicade na více než 16 týdnů, existuje zvýšené riziko alergických reakcí při opětovném zahájení užívání přípravku Remicade.
Infekce
Sdělte svému lékaři, pokud máte „infekci, a to i velmi malou, před podáním přípravku Remicade
- Informujte svého lékaře před podáním přípravku Remicade, pokud jste žili nebo cestovali do „oblasti, kde jsou běžné infekce nazývané histoplazmóza, kokcidioidomykóza nebo blastomykóza. Tyto infekce jsou způsobeny specifickými druhy hub, které mohou postihnout plíce nebo jiné části tělo. tělo
- Při léčbě přípravkem Remicade můžete být náchylnější k infekcím. Pokud je vám 65 nebo více let, máte vyšší riziko
- Tyto infekce mohou být závažné a zahrnují tuberkulózu, infekce způsobené viry, houbami nebo bakteriemi nebo jiné oportunní infekce a sepse, které mohou být ve vzácných případech život ohrožující.
Okamžitě informujte svého lékaře, pokud během léčby přípravkem Remicade zaznamenáte jakékoli příznaky infekce. Mezi příznaky patří horečka, kašel, příznaky podobné chřipce, nevolnost, červená nebo velmi horká kůže, rány nebo problémy se zuby. Váš lékař vám může doporučit dočasné vysazení přípravku Remicade.
Tuberkulóza (TBC)
- Je velmi důležité, abyste informoval svého lékaře, pokud jste někdy měli TBC nebo jste byli v blízkém kontaktu s lidmi, kteří měli nebo mají TBC.
- Váš lékař provede testy, aby zjistil, zda máte tuberkulózu. U pacientů léčených přípravkem Remicade bylo hlášeno několik případů tuberkulózy, ve vzácných případech dokonce u pacientů, kteří byli léčeni léky na TBC. Lékař zaznamená tyto testy na kartu pacienta
- Pokud si váš lékař myslí, že vám hrozí tuberkulóza, můžete být před podáním přípravku Remicade léčeni léky na tuberkulózu.
Okamžitě informujte svého lékaře, pokud si během užívání přípravku Remicade všimnete jakýchkoli známek tuberkulózy. Mezi příznaky patří přetrvávající kašel, hubnutí, pocit únavy, horečka, noční pocení.
Virus hepatitidy B (HBV)
- Před podáním přípravku Remicade informujte svého lékaře, pokud jste nosičem nebo máte nebo jste měl (a) hepatitidu B
- Informujte svého lékaře, pokud si myslíte, že byste mohli být vystaveni riziku nákazy hepatitidou B.
- Měl by lékař vyhodnotit, zda máte hepatitidu B? Léčba blokátory TNF, jako je Remicade, může způsobit reaktivaci viru hepatitidy B u pacientů s tímto virem, což v některých případech může způsobit smrt.
Srdeční problémy
- Informujte svého lékaře, pokud máte nějaké srdeční problémy, jako je mírné srdeční selhání
- Váš lékař bude pečlivě sledovat funkci vašeho srdce.
Okamžitě informujte svého lékaře, pokud během léčby přípravkem Remicade zaznamenáte nové nebo zhoršující se příznaky srdečního selhání. Mezi příznaky patří dušnost nebo otoky nohou.
Rakovina a lymfom
- Před podáním přípravku Remicade informujte svého lékaře, pokud máte nebo jste někdy měl lymfom (typ rakoviny krve) nebo jiné druhy rakoviny
- Pacienti s těžkou revmatoidní artritidou, kteří dlouhodobě trpí touto nemocí, mohou mít vyšší než průměrné riziko vzniku lymfomu.
- Děti a dospělí užívající přípravek Remicade mohou mít zvýšené riziko vzniku lymfomu nebo jiného druhu rakoviny.
- U některých pacientů, kteří byli léčeni blokátory TNF, včetně přípravku Remicade, se vyvinul vzácný typ rakoviny zvaný hepatosplenický T-buněčný lymfom.Většina těchto pacientů byla dospívající nebo mladí dospělí muži a většina měla Crohnovu nebo ulcerózní kolitidu. Tento typ rakoviny je obvykle smrtelný. Téměř všichni pacienti byli kromě blokátorů TNF léčeni také léky zvanými azathioprin nebo 6-merkaptopurin.
- U některých pacientů léčených infliximabem se vyvinuly určité typy rakoviny kůže. Pokud během léčby nebo po ní zaznamenáte jakoukoli změnu vzhledu kůže nebo výrůstky na kůži, sdělte to prosím svému lékaři.
Plicní onemocnění nebo silné kouření
- Informujte svého lékaře, pokud máte plicní onemocnění zvané chronická obstrukční plicní nemoc (CHOPN) nebo pokud silně kouříte, než vám bude podán přípravek Remicade
- Pacienti s CHOPN a silní kuřáci mohou mít při léčbě přípravkem Remicade zvýšené riziko rakoviny.
Onemocnění nervového systému
- Před podáním přípravku Remicade informujte svého lékaře, pokud máte nebo jste někdy měl problém s nervovým systémem. Patří sem roztroušená skleróza, Guillain-Barrého syndrom, záchvaty nebo diagnóza „optické neuritidy“.
Okamžitě informujte svého lékaře, pokud během užívání přípravku Remicade zaznamenáte příznaky nervového onemocnění. Příznaky zahrnují změny vidění, slabost paží a nohou, necitlivost nebo brnění v jakékoli části těla.
Abnormální kožní otvory
- Před podáním přípravku Remicade informujte svého lékaře, pokud máte neobvyklé kožní otvory (píštěle).
Očkování
- Informujte svého lékaře, pokud jste byli nedávno očkováni nebo plánujete očkování podstoupit
- Během léčby přípravkem Remicade nesmíte dostat žádné vakcíny
- Některá očkování mohou způsobit infekce. Pokud jste dostávala přípravek Remicade během těhotenství, může mít vaše dítě zvýšené riziko nákazy touto infekcí přibližně šest měsíců po poslední dávce podané během těhotenství. Je důležité informovat svého pediatra a další zdravotnické pracovníky o používání přípravku Remicade, aby se mohl rozhodnout kdy by vaše dítě mělo dostat jakoukoli vakcínu.
Infekční terapeutická činidla
- Poraďte se svým lékařem, pokud jste v nedávné době užívali nebo plánujete léčbu infekčním terapeutickým činidlem (jako je BCG instilace používaná k léčbě rakoviny).
Zubní operace nebo postupy
- Informujte svého lékaře, pokud se chystáte podstoupit nějaké zubní ošetření nebo ošetření
- Sdělte chirurgovi nebo zubaři provádějícímu zákrok, že jste léčeni přípravkem Remicade, a to ukázáním karty pacienta.
Děti a dospívající
Výše uvedené informace platí také pro děti a mladistvé. Dále:
- u některých dětí a dospívajících pacientů, kteří užívali léky blokující TNF, jako je Remicade, se vyvinula rakovina, včetně neobvyklých typů, která byla někdy smrtelná.
- Ve srovnání s dospělými se u více dětí užívajících přípravek Remicade objevily infekce
- Před zahájením léčby přípravkem Remicade by děti měly dostat doporučené očkování.
Pokud si nejste jisti, zda se na vás vztahuje některý z výše uvedených stavů, obraťte se před podáním přípravku Remicade na svého lékaře.
Interakce Které léky nebo potraviny mohou změnit účinek přípravku Remicade
Pacienti se zánětlivým onemocněním již užívají léky k léčbě onemocnění. Tyto léky mohou způsobit nežádoucí účinky. Váš lékař vám poradí, jaké další léky byste měli v průběhu léčby přípravkem Remicade nadále užívat.
Informujte svého lékaře, pokud užíváte nebo jste v nedávné době užíval jiné léky, včetně jakýchkoli jiných léků k léčbě Crohnovy choroby, ulcerózní kolitidy, revmatoidní artritidy, ankylozující spondylitidy, psoriatické artritidy nebo psoriázy nebo léků, které dostanete bez lékařského předpisu, jako jsou vitamíny a byliny léky.
Zejména informujte svého lékaře, pokud užíváte některý z těchto léků:
- Léky, které ovlivňují imunitní systém
- Kineret (anakinra). Remicade a Kineret se nesmí podávat společně
- Orencia (abatacept). Remicade a Orencia se nesmí podávat společně.
Pokud si nejste jisti, zda se na vás vztahuje některý z výše uvedených stavů, obraťte se před podáním přípravku Remicade na svého lékaře.
Varování Je důležité vědět, že:
Těhotenství, kojení a plodnost
- Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Remicade se během těhotenství nedoporučuje
- Během léčby přípravkem Remicade a po dobu nejméně 6 měsíců po ukončení léčby se musíte vyhnout otěhotnění.Ujistěte se, že během této doby používáte vhodnou antikoncepci.
- Během léčby přípravkem Remicade nebo 6 měsíců po poslední léčbě přípravkem Remicade nekojte
- Pokud jste dostali přípravek Remicade během těhotenství, může mít vaše dítě zvýšené riziko infekce. Je důležité informovat svého pediatra a další zdravotnické pracovníky o tom, jak přípravek Remicade používáte, než vaše dítě dostane jakoukoli vakcínu (další informace viz část o očkování ).
Řízení dopravních prostředků a obsluha strojů
Je nepravděpodobné, že by Remicade ovlivňoval vaši schopnost řídit nebo obsluhovat stroje. Pokud se po léčbě Remicade cítíte unavení nebo se necítíte dobře, neměli byste řídit ani používat žádné nástroje nebo stroje.
Dávka, způsob a doba podání Jak používat Remicade: Dávkování
Jak se přípravek Remicade podává
- Remicade vám podá lékař nebo zdravotní sestra
- Váš lékař nebo zdravotní sestra připraví injekční roztok Remicade
- Roztok Remicade bude injekčně podáván pomalu (po dobu 2 hodin) do žíly, obvykle do paže. Tento postup se nazývá „intravenózní infuze“ nebo kapání. Po třetím ošetření se váš lékař může rozhodnout podat vám přípravek Remicade po dobu 1 hodiny
- Během podávání přípravku Remicade a po dobu 1–2 hodin poté budete sledováni.
Kolik přípravku Remicade se podává
- Váš lékař stanoví dávku (v mg) a interval mezi dávkami přípravku Remicade. To bude záviset na vaší nemoci, hmotnosti a reakci na léčbu.
- Níže uvedená tabulka ukazuje frekvenci podávání tohoto léku.
Revmatoidní artritida
Obvyklá dávka je 3 mg na každý kg tělesné hmotnosti
Psoriatická artritida, ankylozující spondylitida (Bechtěrevova choroba), psoriáza, ulcerózní kolitida a Crohnova choroba
Obvyklá dávka je 5 mg na každý kg tělesné hmotnosti.
Použití u dětí a dospívajících
Remicade je určen pouze k použití u dětí s Crohnovou chorobou nebo ulcerózní kolitidou. Těmto dětem musí být 6 let nebo starší.
Předávkování Co dělat, když jste užil příliš mnoho přípravku Remicade
Pokud dostanete více přípravku Remicade, než potřebujete
Jelikož vám tento lék podává lékař nebo zdravotní sestra, je nepravděpodobné, že byste dostali příliš mnoho. Nejsou známy žádné vedlejší účinky předávkování přípravkem Remicade.
Jestliže zapomenete nebo vynecháte infuzi „Remicade“
Pokud zapomenete nebo zmeškáte schůzku ke správě Remicade, proveďte další schůzku co nejdříve.
Máte -li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře
Nežádoucí účinky Jaké jsou vedlejší účinky přípravku Remicade
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Většina těchto účinků je mírná až střední. U některých pacientů se však mohou vyskytnout závažné nežádoucí účinky a vyžadují lékařské ošetření. Nežádoucí účinky se mohou objevit také po ukončení léčby přípravkem Remicade.
Okamžitě informujte svého lékaře, pokud zaznamenáte některý z následujících nežádoucích účinků:
- Známky alergické reakce, jako je otok obličeje, rtů, úst nebo hrdla, který může způsobit potíže s polykáním nebo dýcháním, vyrážka, kopřivka, otok rukou, nohou nebo kotníků. Alergická reakce se může objevit do 2 hodin po injekci nebo později. Další příznaky alergické reakce, které se mohou objevit až 12 dní po injekci, zahrnují bolesti svalů, horečku, bolest kloubů nebo čelistí, bolest v krku nebo bolest v krku.
- Známky srdečního problému, jako je dušnost, otoky nohou nebo změny srdečního tepu
- Známky infekce (včetně tuberkulózy), jako je horečka, pocit únavy, kašel (přetrvávající), dušnost, příznaky podobné chřipce, úbytek na váze, noční pocení, průjem, rány, problémy se zuby nebo pálení při močení
- Známky plicního problému, jako je kašel, potíže s dýcháním nebo tlak na hrudi
- Známky neurologických problémů (včetně problémů s očima), jako jsou záchvaty, brnění nebo necitlivost v jakékoli části těla, slabost paží nebo nohou, změny vidění, jako je dvojité vidění nebo jiné oční problémy
- Známky problému s játry, jako je zežloutnutí kůže nebo očí, tmavě hnědá moč nebo bolest v pravé horní části žaludku, horečka
- Známky poruchy imunitního systému, zvané lupus, jako bolest kloubů nebo vyrážka na tvářích nebo pažích, oblasti citlivé na slunce
- Známky poklesu počtu krvinek, jako je přetrvávající horečka, častější krvácení nebo tvorba modřin nebo bledý vzhled.
Pokud zaznamenáte některý z výše popsaných příznaků, ihned to sdělte svému lékaři.
Velmi časté nežádoucí účinky (postihují více než 1 z 10 pacientů)
- Bolest žaludku, malátnost
- Virové infekce, jako je herpes nebo chřipka
- Infekce horních cest dýchacích, jako je zánět vedlejších nosních dutin
- Bolest hlavy
- Nežádoucí účinek v důsledku infuze
- Bolest.
Časté nežádoucí účinky (postihují 1 až 10 uživatelů ze 100)
- Změny funkce jater, zvýšení jaterních enzymů (pozorováno v krevních testech)
- Infekce plic nebo hrudníku, jako je bronchitida nebo pneumonie
- Potíže s dýcháním nebo bolest při dýchání, bolest na hrudi
- Krvácení do žaludku nebo střev, průjem, poruchy trávení, pálení žáhy, zácpa
- Vyrážka připomínající kopřivku, svědivá vyrážka nebo suchá kůže
- Problémy s rovnováhou nebo pocit závratě
- Horečka, zvýšené pocení
- Problémy s oběhem, jako je nízký nebo vysoký krevní tlak
- Modřiny, zrudnutí nebo krvácení z nosu, horká, zarudlá kůže (zarudnutí)
- Pocit únavy nebo slabosti
- Bakteriální infekce, jako je generalizovaná infekce, absces nebo infekce hlubokých vrstev kůže (celulitida)
- Krevní problémy, jako je anémie nebo nízký počet bílých krvinek
- Zvětšené lymfatické uzliny
- Deprese, poruchy spánku
- Oční problémy, včetně červených očí a infekcí
- Rychlý srdeční tep (tachykardie) nebo palpitace
- Bolest kloubů, svalů nebo zad
- Infekce močových cest
- Psoriáza, kožní problémy jako ekzém a vypadávání vlasů
- Reakce v místě vpichu, jako je bolest, otok, zarudnutí nebo svědění
- Zimnice, nahromadění tekutiny pod kůží způsobující otok
- Necitlivost nebo pocit brnění.
Méně časté nežádoucí účinky (postihují 1 až 10 uživatelů z 1 000)
- Špatné prokrvení, otok žíly
- Kožní problémy, jako jsou puchýře, bradavice, neobvyklé zabarvení nebo pigmentace kůže nebo oteklé rty
- Závažné alergické reakce (např. Anafylaxe), porucha imunitního systému zvaná lupus, alergické reakce na cizí bílkoviny
- Rány, které se pomalu hojí
- Otok jater (hepatitida) nebo žlučník (žlučník), poškození jater
- Rozptýlení, podrážděnost, zmatenost, nervozita
- Oční problémy zahrnující rozmazané nebo snížené vidění, oteklé oči nebo styly
- Nové nebo zhoršené srdeční selhání, pomalá srdeční frekvence
- Mdloby
- Křeče, nervové poruchy
- Perforace nebo zablokování střev, bolest žaludku nebo křeče
- Otok slinivky břišní (pankreatitida)
- Plísňové infekce, jako je kvasinková infekce
- Plicní problémy (jako edém)
- Nadměrná tekutina kolem plic (pleurální výpotek)
- Infekce ledvin
- Nízký počet krevních destiček, nadměrný počet bílých krvinek
- Infekce v pochvě.
Vzácné nežádoucí účinky (postihují 1 až 10 uživatelů z 10 000)
- Typ rakoviny krve (lymfom)
- Špatný přísun kyslíku do orgánů krví, problémy s cirkulací, jako je zúžení cévy
- Zánět membrány, která lemuje mozek (meningitida)
- Infekce v důsledku oslabeného imunitního systému
- Infekce hepatitidy B, pokud jste v minulosti měli hepatitidu B? Otok nebo růst abnormálních tkání
- Otok malých cév (vaskulitida)? Imunologické poruchy, které mohou postihnout plíce, kůži a lymfatické uzliny (jako je sarkoidóza)
- Nedostatek zájmu nebo emocí
- Závažné kožní problémy, jako je toxická epidermální nekrolýza, Steven-Johnsonův syndrom nebo multiformní erytém, kožní problémy, jako jsou vředy
- Závažné poruchy nervového systému, jako je příčná myelitida, onemocnění podobné roztroušené skleróze, optická neuritida a Guillain-Barrého syndrom
- Tekutina v membráně, která lemuje srdce (perikardiální výpotek)
- Závažné plicní problémy (jako je intersticiální pneumonie)
- Melanom (typ rakoviny kůže).
Další nežádoucí účinky (frekvence není známa)
- Rakovina u dětí a dospělých
- Vzácný nádor krve, který postihuje hlavně mladé lidi (hepatosplenický T-buněčný lymfom)
- Jaterní nedostatečnost
- Karcinom z Merkelových buněk (typ rakoviny kůže)
- Zhoršení stavu zvaného dermatomyozitida (vypadá jako „vyrážka, která doprovází svalovou slabost).
Další nežádoucí účinky u dětí a dospívajících
Děti, které užívaly Remicade na Crohnovu chorobu, vykazovaly určité rozdíly ve vedlejších účincích ve srovnání s dospělými, kteří užívali Remicade na Crohnovu chorobu.
Nejčastějšími nežádoucími účinky u dětí byly: nízký počet červených krvinek (anémie), krev ve stolici, nízký počet bílých krvinek (leukopenie), zrudnutí nebo zarudnutí (návaly horka), virové infekce, nízký počet neutrofilů (neutropenie), které jsou bílé krvinky, které bojují s infekcí, zlomeninou kostí, bakteriální infekcí a alergickými reakcemi dýchacích cest.
Hlášení nežádoucích účinků
Pokud se u vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. To se týká i všech možných nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. poskytnout více informací o bezpečnosti tohoto léku.
Expirace a retence
Remicade budou obvykle uchovávány zdravotnickými pracovníky. Pokud byste to potřebovali, podrobnosti o uchování jsou následující:
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku a krabičce za „EXP“. Datum exspirace se vztahuje k poslednímu dni uvedeného měsíce.
- Uchovávejte v chladničce (2 ° C - 8 ° C).
- Tento léčivý přípravek lze také uchovávat v původním obalu mimo chladničku do maximálně 25 ° C po dobu jednoho období až šesti měsíců. V této situaci by neměl být znovu uchováván v chladničce. Do pole napište nové datum vypršení platnosti včetně dne / měsíce / roku. Zlikvidujte tento lék, pokud není použit do nového data expirace nebo do data exspirace vytištěného na krabičce, podle toho, co nastane dříve.
- Když je přípravek Remicade připraven k infuzi, doporučuje se jej použít co nejdříve (do 3 hodin). Pokud je však roztok připraven za zcela zárodečných podmínek, může být uchováván v chladničce po dobu 24 hodin při teplotě mezi 2 ° C. ° C a 8 ° C
- Nepoužívejte tento přípravek, pokud má změněnou barvu nebo obsahuje částice.
Co přípravek Remicade obsahuje
- Léčivou látkou je infliximab. Jedna injekční lahvička obsahuje infliximabum 100 mg. Po přípravě každý ml obsahuje 10 mg infliximabu.
- Dalšími složkami jsou sacharóza, polysorbát 80, jednosytný fosforečnan sodný a hydrogenfosforečnan sodný.
Jak Remicade vypadá a obsah balení
Remicade je dodáván ve skleněné lahvičce obsahující prášek pro koncentrát pro infuzní roztok. Prášek se skládá z lyofilizovaných bílých granulí.
Remicade je k dispozici v balení po 1, 2, 3, 4 nebo 5 injekčních lahvičkách. Na trhu nemusí být všechny velikosti balení
Zdroj příbalové informace: AIFA (Italská agentura pro léčivé přípravky). Obsah zveřejněný v lednu 2016. Přítomné informace nemusí být aktuální.
Abyste měli přístup k nejaktuálnější verzi, doporučujeme navštívit webovou stránku AIFA (Italská agentura pro léčivé přípravky). Prohlášení a užitečné informace.
01.0 NÁZEV LÉČIVÉHO PŘÍPRAVKU
REMICADE 100 MG PRÁŠEK PRO KONCENTRÁT PRO ROZTOK K INFUZI
02.0 KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje infliximabum 100 mg. Infliximab je chimérická lidská myší IgG1 monoklonální protilátka produkovaná v myších hybridomových buňkách technologií rekombinantní DNA. Po rekonstituci obsahuje každý ml 10 mg infliximabu.
Úplný seznam pomocných látek viz bod 6.1.
03.0 LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok.
Prášek se skládá z lyofilizovaných bílých granulí.
04.0 KLINICKÉ INFORMACE
04.1 Terapeutické indikace
Revmatoidní artritida
Remicade v kombinaci s methotrexátem je indikován ke snížení známek a příznaků a zlepšení tělesných funkcí:
• dospělí pacienti s aktivním onemocněním, u nichž byla reakce na onemocnění modifikující antirevmatika (DMARD), včetně methotrexátu, nedostatečná.
• dospělí pacienti s těžkým, aktivním a progresivním onemocněním, kteří nebyli dříve léčeni methotrexátem nebo jinými DMARD.
Snížení rychlosti progrese poškození kloubů bylo prokázáno radiografickým hodnocením v této populaci pacientů (viz bod 5.1).
Crohnova choroba u dospělých
Remicade je indikován pro:
• léčba středně těžké až těžké aktivní Crohnovy choroby u dospělých pacientů, kteří nereagovali navzdory úplné a adekvátní léčbě kortikosteroidy a / nebo imunosupresivy; nebo u pacientů, kteří netolerují nebo mají zdravotní kontraindikace pro výše uvedené terapie.
• léčba aktivní fistulizující Crohnovy choroby u dospělých pacientů, kteří nereagovali navzdory úplnému a adekvátnímu průběhu léčby konvenční léčbou (včetně antibiotik, drenáže a imunosupresivní terapie).
Crohnova choroba u dětí
Remicade je indikován k léčbě závažné aktivní Crohnovy choroby u dětí a dospívajících ve věku 6-17 let, kteří nereagovali na konvenční léčbu kortikosteroidy, imunomodulátorem a primární nutriční terapií, nebo u pacientů, kteří netolerují nebo mají kontraindikace pro výše uvedené terapie. Remicade byl studován pouze v kombinaci s konvenční imunosupresivní terapií.
Ulcerózní kolitida
Remicade je indikován k léčbě středně těžké až těžké aktivní ulcerózní kolitidy u dospělých pacientů, kteří dostatečně nereagovali na konvenční terapii zahrnující kortikosteroidy a 6-merkaptopurin (6-MP) nebo azathioprin (AZA), nebo kteří jsou nesnášenliví nebo u nichž existuje zdravotní kontraindikace těchto terapií.
Dětská ulcerózní kolitida
Remicade je indikován k léčbě těžké aktivní ulcerózní kolitidy u dětí a mladistvých ve věku 6 až 17 let, kteří dostatečně nereagovali na konvenční léčbu zahrnující kortikosteroidy a 6-MP nebo AZA, nebo kteří trpí nesnášenlivostí nebo u nichž existuje lékařská kontraindikace těchto terapií.
Ankylozující spondylitida
Remicade je indikován k léčbě těžké aktivní ankylozující spondylitidy u dospělých pacientů, kteří dostatečně nereagovali na konvenční terapie.
Psoriatická artritida
Remicade je indikován k léčbě aktivní a progresivní psoriatické artritidy u dospělých pacientů, u nichž byla reakce na předchozí léčbu DMARD nedostatečná.
Remicade by měl být podáván:
• ve spojení s methotrexátem
• nebo jednotlivě u pacientů, kteří netolerují methotrexát nebo u kterých je kontraindikován
Bylo prokázáno, že přípravek Remicade zlepšuje fyzické funkce u pacientů s psoriatickou artritidou a snižuje rychlost progrese poškození periferních kloubů měřeno rentgenovým zářením u pacientů se symetrickými polyartikulárními podtypy onemocnění (viz bod 5.1).
Lupénka
Remicade je indikován k léčbě středně těžké až těžké ložiskové psoriázy u dospělých pacientů, u nichž selhala nebo jsou kontraindikováni nebo kteří nesnášeli jinou systémovou léčbu včetně cyklosporinu, methotrexátu nebo PUVA (viz bod 5.1).
04.2 Dávkování a způsob podání
Léčbu přípravkem Remicade by měl zahájit a sledovat odborný lékař se zkušenostmi s diagnostikou a léčbou revmatoidní artritidy, zánětlivého onemocnění střev, ankylozující spondylitidy, psoriatické artritidy nebo psoriázy. Remicade musí být podáván intravenózně. Infuze přípravku Remicade by měl podávat kvalifikovaný zdravotnický personál vyškolený v rozpoznávání jakýchkoli problémů souvisejících s infuzí.Pacientům léčeným přípravkem Remicade by měla být poskytnuta příbalová informace a výstražná karta pacienta.
Během léčby přípravkem Remicade by mělo být optimalizováno používání dalších souběžných terapií, jako jsou kortikosteroidy a imunosupresiva.
Dávkování
Dospělí (≥ 18 let)
Revmatoidní artritida
Intravenózní infuze 3 mg / kg následovaná dalšími infuzemi 3 mg / kg ve 2. a 6. týdnu po první infuzi, poté každých 8 týdnů.
Remicade musí být podáván současně s methotrexátem.
Dostupné údaje naznačují, že klinické odpovědi je obvykle dosaženo do 12 týdnů od zahájení léčby. Pokud má pacient nedostatečnou odpověď nebo odpověď po tomto období ztratí, lze uvažovat o postupném zvyšování dávky o 1,5 mg. / Kg, a to až do maximální 7,5 mg / kg, každých 8 týdnů. Alternativně lze zvážit podávání 3 mg / kg každé 4 týdny. Pokud je dosaženo adekvátní odpovědi, léčba by měla pokračovat. pacienti se zvolenou dávkou nebo frekvencí. Je třeba pečlivě zvážit, zda pokračovat v léčbě u pacientů, kteří během prvních 12 týdnů léčby nebo po úpravě dávky nevykazují žádný terapeutický přínos.
Středně těžká až těžká aktivní Crohnova choroba
5 mg / kg podáno jako intravenózní infuze následovaná další infuzí 5 mg / kg 2 týdny po první infuzi. Pokud pacient nereaguje na terapii ani po 2 dávkách, nemá být další léčba infliximabem prováděna. Dostupné údaje nepodporují další léčbu infliximabem u pacientů, kteří nejsou pacienty respondenti do 6 týdnů od první infuze.
U reagujících pacientů jsou alternativními řešeními pro pokračující léčbu:
• Údržba: doplňková infuze 5 mg / kg v 6. týdnu po první dávce, po níž následují opakované infuze každých 8 týdnů nebo
• Opětovné podání: infuze 5 mg / kg, pokud příznaky a příznaky onemocnění přetrvávají (viz „Opětovné podání“ a bod 4.4).
Ačkoli chybí srovnávací údaje, omezené údaje u pacientů, kteří zpočátku reagovali na léčbu 5 mg / kg, ale ztratili odpověď, naznačují, že někteří pacienti mohou obnovit odpověď zvýšením dávky (viz bod 5.1). Pokračování léčby by mělo být pečlivě zváženo u pacientů, kteří po úpravě dávky nevykazují žádný terapeutický přínos.
Aktivní fistulizující Crohnova choroba
5 mg / kg podáno formou intravenózní infuze následované dalšími 5 mg / kg infuzí ve 2. a 6. týdnu po první infuzi. Pokud pacient nereaguje ani po 3 dávkách, nemá být další léčba infliximabem prováděna.
U reagujících pacientů jsou alternativními řešeními pro pokračující léčbu:
• Údržba: další infuze 5 mg / kg každých 8 týdnů nebo
• Opakované podání: infuze 5 mg / kg, pokud příznaky a symptomy onemocnění přetrvávají, následovaná infuzí 5 mg / kg každých 8 týdnů (viz „Opakované podání“ a bod 4.4).
Ačkoli chybí srovnávací údaje, omezené údaje u pacientů, kteří zpočátku reagovali na léčbu 5 mg / kg, ale ztratili odpověď, naznačují, že někteří pacienti mohou obnovit odpověď zvýšením dávky (viz bod 5.1). Pokračování léčby by mělo být pečlivě zváženo u pacientů, kteří po úpravě dávky nevykazují žádný terapeutický přínos.
U Crohnovy choroby jsou zkušenosti s opětovným podáním, pokud příznaky a symptomy onemocnění přetrvávají, omezené a nejsou k dispozici srovnatelné údaje o poměru rizika a přínosu alternativních řešení pro pokračování léčby.
Ulcerózní kolitida
Intravenózní infuze 5 mg / kg následovaná dalšími 5 mg / kg infuzí ve 2. a 6. týdnu po první infuzi, poté se opakuje každých 8 týdnů.
Dostupné údaje naznačují, že klinické odpovědi je obvykle dosaženo do 14 týdnů od zahájení léčby, tj. Po třech podáních. Pečlivě je třeba zvážit pokračování léčby u pacientů, kteří během této doby nereagují.
Ankylozující spondylitida
Intravenózní infuze 5 mg / kg následovaná dalšími infuzí 5 mg / kg ve 2. a 6. týdnu po první infuzi, poté opakovaná po 6 až 8 týdnech. Pokud pacient neodpoví do 6 týdnů (tj. Po 2 dávkách), neměl by dostávat žádnou další léčbu infliximabem.
Psoriatická artritida
Intravenózní infuze 5 mg / kg následovaná dalšími infuzí 5 mg / kg ve 2. a 6. týdnu po první infuzi, poté se opakuje každých 8 týdnů.
Lupénka
Intravenózní infuze 5 mg / kg následovaná dalšími infuzí 5 mg / kg ve 2. a 6. týdnu po první infuzi, poté se opakuje každých 8 týdnů. Pokud pacient neodpoví do 14 týdnů (tj. Po 4 dávkách), nemá být další léčba infliximabem prováděna.
Opětovné podání u Crohnovy choroby a revmatoidní artritidy
Pokud se příznaky a příznaky onemocnění opakují, lze Remicade znovu podat do 16 týdnů od poslední infuze. V klinických studiích byly opožděné reakce z přecitlivělosti „neobvyklé“ a vyskytovaly se po intervalech bez podání přípravku Remicade. Méně než 1 rok (viz body 4.4 a 4.8). Bezpečnost a účinnost opětovného podání nebyla stanovena po více než 16 týdnech bez podání přípravku Remicade. To platí jak pro pacienty s Crohnovou chorobou, tak pro pacienty s revmatoidní artritidou.
Opětovné podání u ulcerózní kolitidy
Bezpečnost a účinnost opakovaného podávání v intervalech jiných než 8 týdnů nebyla stanovena (viz body 4.4 a 4.8).
Opětovné podání ankylozující spondylitidy
Bezpečnost a účinnost opakovaných podání jiných než těch, která byla podávána v intervalu 6 až 8 týdnů, nebyla stanovena (viz body 4.4 a 4.8).
Opětovné podání u psoriatické artritidy
Bezpečnost a účinnost opakovaného podávání v intervalech jiných než 8 týdnů nebyla stanovena (viz body 4.4 a 4.8).
Opětovné podání u psoriázy
„Omezené zkušenosti s psoriázou vyplývající z opakované léčby jednou dávkou přípravku Remicade po intervalu 20 týdnů naznačují„ sníženou účinnost a „vyšší výskyt mírných až středně závažných reakcí na infuzi“ ve srovnání s počátečním indukčním režimem (viz. bod 5.1).
„Omezené zkušenosti s opakovanou léčbou po zhoršení onemocnění prostřednictvím reindukčního režimu naznačují“ vysoký výskyt reakcí na infuzi, včetně závažných, ve srovnání s reakcemi po 8 týdnech udržovací léčby (viz bod 4.8).
Opětovné podání v různých indikacích
V případě, že je udržovací léčba přerušena a je třeba léčbu znovu zahájit, nedoporučuje se použití režimu opětné indukce (viz bod 4.8) .V této situaci by měla být léčba přípravkem Remicade znovu zahájena jako jednotlivá dávka a poté udržovací dávka podle výše popsaných doporučení.
Starší pacienti (≥ 65 let)
Nebyly provedeny žádné specifické studie s přípravkem Remicade u starších pacientů. V klinických studiích nebyly pozorovány žádné podstatné rozdíly v clearance nebo distribučním objemu související s věkem.
Úprava dávky není nutná (viz bod 5.2). Další informace o bezpečnosti přípravku Remicade u starších pacientů viz body 4.4 a 4.8.
Porucha funkce ledvin a / nebo jater
Remicade nebyl u těchto populací pacientů studován. Nelze učinit doporučení pro dávku (viz bod 5.2).
Pediatrická populace
Crohnova choroba (6 - 17 let)
Dávka 5 mg / kg podaná intravenózní infuzí, po níž následují následné infuze dávek 5 mg / kg ve 2 a 6 týdnech po první infuzi a poté každých 8 týdnů. Dostupné údaje nepodporují další léčbu infliximabem u dětí a dospívajících, kteří během prvních 10 týdnů léčby nereagují (viz bod 5.1).
Někteří pacienti mohou vyžadovat kratší interval mezi dávkami, aby si udrželi klinický prospěch, zatímco jiným může stačit delší interval mezi dávkami. U pacientů, u nichž byl časový interval mezi dávkami snížen na méně než 8 týdnů, může být zvýšené riziko nežádoucích účinků.Pokračování léčby se zkráceným intervalem by mělo být pečlivě zváženo u pacientů, u nichž není prokázán terapeutický přínos. Po změně časový interval mezi dávkami.
Bezpečnost a účinnost přípravku Remicade u dětí s Crohnovou chorobou do 6 let nebyla studována.V současnosti dostupné farmakokinetické údaje jsou popsány v bodě 5.2, ale u dětí mladších 6 let nelze učinit žádná doporučení ohledně dávkování.
Ulcerózní kolitida (6 - 17 let)
Dávka 5 mg / kg podaná intravenózní infuzí, po níž následují následné infuze dávek 5 mg / kg ve 2 a 6 týdnech po první infuzi a poté každých 8 týdnů. Dostupné údaje nepodporují další léčbu infliximabem u pediatrických pacientů, kteří během prvních 8 týdnů léčby nereagují (viz bod 5.1).
Bezpečnost a účinnost přípravku Remicade u dětí s ulcerózní kolitidou mladších 6 let nebyla studována. V současnosti dostupné farmakokinetické údaje jsou popsány v bodě 5.2, ale u dětí mladších 6 let nelze učinit žádná doporučení ohledně dávkování.
Lupénka
Bezpečnost a účinnost přípravku Remicade u dětí a dospívajících mladších 18 let v indikaci psoriázy nebyla stanovena. V současné době jsou dostupná data popsána v bodě 5.2, ale nelze učinit žádná doporučení ohledně dávkování.
Juvenilní idiopatická artritida, psoriatická artritida a ankylozující spondylitida
Bezpečnost a účinnost přípravku Remicade u dětí a mladistvých do 18 let v indikacích juvenilní idiopatická artritida, psoriatická artritida a ankylozující spondylitida nebyla stanovena. Aktuálně dostupné údaje jsou popsány v bodě 5.2, ale nelze učinit žádná doporučení ohledně dávkování.
Juvenilní revmatoidní artritida
Bezpečnost a účinnost přípravku Remicade u dětí a mladistvých do 18 let v indikaci juvenilní revmatoidní artritidy nebyla stanovena. V současné době jsou dostupné údaje popsány v bodech 4.8 a 5.2, ale nelze je provést. Doporučení ohledně dávkování.
Porucha funkce ledvin a / nebo jater
Remicade nebyl u těchto populací pacientů studován. Nelze učinit doporučení pro dávku (viz bod 5.2).
Způsob podání
Remicade by měl být podáván intravenózně po dobu 2 hodin. Všichni pacienti léčení přípravkem Remicade by měli být po dobu nejméně 1–2 hodin po infuzi sledováni kvůli akutním reakcím souvisejícím s infuzí. Mělo by být k dispozici nouzové vybavení, jako je adrenalin, antihistaminika, kortikosteroidy a umělý respirátor. Pacienti mohou být předem ošetřeni například antihistaminikem, hydrokortisonem a / nebo paracetamolem a rychlost infuze může být zpomalena, aby se snížilo riziko infuze- související reakce, zvláště pokud se reakce související s infuzí vyskytly již dříve (viz bod 4.4).
Infuze zkráceny v indikacích pro dospělé
U pečlivě vybraných dospělých pacientů, kteří tolerovali alespoň 3 úvodní 2hodinové infuze Remicade (indukční fáze) a kteří dostávají udržovací terapii, podávání následných infuzí po dobu nejméně 1 hodiny Pokud reakce na infuzi spojená se zkrácenou infuzí nastane, může být u budoucích infuzí zvažována nižší rychlost infuze, pokud by léčba pokračovala. Zkrácené infuze v dávkách> 6 mg / kg nebyly studovány (viz bod 4.8).
Pokyny k přípravě a podání viz bod 6.6.
04.3 Kontraindikace
Pacienti s anamnézou přecitlivělosti na infliximab (viz bod 4.8), na jiné myší proteiny nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Pacienti s tuberkulózou nebo jinými závažnými infekcemi, jako je sepse, abscesy a oportunní infekce (viz bod 4.4).
Pacienti se středně těžkým až těžkým srdečním selháním (NYHA - New York Heart Association - třída III / IV) (viz body 4.4 a 4.8).
04.4 Zvláštní upozornění a vhodná opatření pro použití
Aby se zlepšila sledovatelnost biologických léčivých přípravků, ochranná známka a číslo šarže podaného přípravku by měly být jasně zaznamenány (nebo označeny) v záznamu pacienta.
Infuzní reakce a přecitlivělost
Infliximab byl spojován s akutními reakcemi souvisejícími s infuzí, včetně anafylaktického šoku a opožděných reakcí z přecitlivělosti (viz bod 4.8).
Akutní reakce na infuzi včetně anafylaktických reakcí se mohou objevit během (do několika sekund) nebo do hodin po infuzi. Pokud dojde k akutním reakcím na infuzi, musí být infuze okamžitě zastavena. Mělo by být k dispozici nouzové vybavení, jako je adrenalin, antihistaminika, kortikosteroidy a umělý ventilátor.
Mohou se vyvinout protilátky proti infliximabu, které byly spojeny se zvýšenou frekvencí reakcí na infuzi. Nízká rychlost reakcí na infuzi byla závažnými alergickými reakcemi a byla také pozorována souvislost mezi tvorbou protilátek proti infliximabu a sníženou odpovědí. Souběžné podávání imunomodulátorů bylo spojeno s nižším výskytem protilátek proti infliximabu a snížením frekvence reakcí na infuzi Účinek souběžné imunomodulační terapie byl u epizodicky léčených pacientů intenzivnější než u pacientů léčených udržovací léčbou. U pacientů, kteří přerušili léčbu imunosupresivy před nebo během léčby přípravkem Remicade, je zvýšené riziko vzniku těchto protilátek. Protilátky proti infliximabu nelze vždy detekovat ve vzorcích séra. Pokud dojde k závažným reakcím, je třeba zahájit symptomatickou léčbu a dále nepodávat další infuze přípravku Remicade (viz bod 4.8.).
V klinických studiích byly hlášeny opožděné reakce přecitlivělosti. Dostupné údaje naznačují zvýšené riziko opožděné přecitlivělosti na prodloužení časových intervalů bez podávání přípravku Remicade.Pacientům by mělo být doporučeno, aby v případě opožděné nežádoucí příhody okamžitě kontaktovali svého lékaře (viz bod 4.8) .Pokud jsou pacienti po delší době ustoupeni období by měli být pečlivě sledováni z hlediska známek a příznaků opožděné přecitlivělosti.
Infekce
Před léčbou, během léčby a po léčbě přípravkem Remicade by měli být pacienti pečlivě sledováni na přítomnost infekcí včetně tuberkulózy. Vzhledem k tomu, že eliminace infliximabu může trvat až šest měsíců, mělo by v tomto období pokračovat sledování.Pokud se u pacienta rozvinou závažné infekce nebo sepse, další léčba přípravkem Remicade by neměla být prováděna.
Při použití přípravku Remicade u pacientů s chronickou infekcí nebo s anamnézou rekurentních infekcí, včetně souběžné léčby imunosupresivy, je nutná opatrnost.Pacienti by měli být náležitě informováni o nutnosti vyhnout se expozici potenciálním rizikovým faktorům infekcí.
Faktor alfa nádorové nekrózy (TNFα) zprostředkovává zánět a moduluje buněčné imunitní odpovědi Experimentální data ukazují, že TNFα je nezbytný pro řešení intracelulárních infekcí.Klinické zkušenosti ukazují, že imunitní obrana hostitele je u některých pacientů léčených infliximabem narušena.
Je třeba poznamenat, že potlačení TNFα může maskovat příznaky infekce, jako je horečka. Včasné rozpoznání atypických klinických projevů závažných infekcí a typických klinických projevů vzácných a neobvyklých infekcí je zásadní pro minimalizaci zpoždění v diagnostice a léčbě.
Pacienti užívající léky blokující TNF jsou náchylnější k závažným infekcím.
U pacientů léčených infliximabem byla pozorována tuberkulóza, bakteriální infekce, včetně sepse a pneumonie, invazivní houbové, virové a jiné oportunní infekce. Některé z těchto infekcí byly smrtelné; k nejčastěji hlášeným oportunním infekcím s úmrtností> 5% patří penumocystóza, kandidóza, listerióza a aspergilóza.
Pacienti, u kterých se během léčby přípravkem Remicade objeví nová infekce, by měli být pečlivě sledováni a podrobeni důkladnému diagnostickému vyšetření. Podávání přípravku Remicade by mělo být přerušeno, pokud se u pacienta rozvine nová závažná infekce nebo sepse, a měla by být zahájena příslušná antimikrobiální nebo antifungální terapie, dokud se infekce nevyřeší.
Tuberkulóza
U pacientů léčených přípravkem Remicade byly hlášeny případy aktivní tuberkulózy. Je třeba poznamenat, že ve většině těchto případů se jednalo o extrapulmonální tuberkulózu, lokalizovanou i difúzní.
Před zahájením léčby přípravkem Remicade by měli být všichni pacienti vyšetřeni na aktivní i neaktivní („latentní“) tuberkulózu. Toto hodnocení by mělo zahrnovat podrobnou anamnézu včetně osobní anamnézy tuberkulózy nebo možného předchozího kontaktu se zdrojem infekce TBC a předchozích a / nebo souběžných imunosupresivních terapií. U všech pacientů by měly být provedeny příslušné diagnostické testy, jako je kožní test na tuberkulinu a rentgenová vyšetření hrudníku (mohou platit místní směrnice). Doporučuje se, aby byly tyto testy zaznamenány na kartě pacienta, která předepisuje lékařům, kteří jsou upozorněni na riziko falešně negativních výsledků kožních testů na tuberkulinu, zejména u těžce nemocných nebo imunokompromitovaných pacientů.
Pokud je diagnostikována aktivní tuberkulóza, léčba přípravkem Remicade nesmí být zahájena. (viz bod 4.3)
Pokud je podezření na latentní tuberkulózu, je třeba se poradit s lékařem, který má zkušenosti s léčbou tuberkulózy. Ve všech níže popsaných situacích je třeba pečlivě zvážit poměr přínosů a rizik léčby přípravkem Remicade.
Pokud je diagnostikována neaktivní („latentní“) tuberkulóza, měla by být před zahájením léčby přípravkem Remicade podle místních pokynů zahájena antituberkulózní léčba latentní tuberkulózy.
U pacientů, kteří mají mnoho nebo významné rizikové faktory pro tuberkulózu a kteří mají negativní test na latentní tuberkulózu, je třeba před zahájením léčby přípravkem Remicade zvážit antituberkulózní léčbu.
Před zahájením léčby přípravkem Remicade by mělo být také zváženo použití léčby proti tuberkulóze u pacientů s předchozí anamnézou latentní nebo aktivní tuberkulózy, u nichž nelze potvrdit adekvátní průběh léčby.
U pacientů léčených přípravkem Remicade během a po léčbě latentní tuberkulózy byly hlášeny některé případy aktivní tuberkulózy.
Všichni pacienti by měli být poučeni, aby vyhledali lékařskou pomoc, pokud se během léčby přípravkem Remicade nebo po ní objeví příznaky / příznaky naznačující tuberkulózu (např. Přetrvávající kašel, chřadnutí / úbytek na váze, nízká horečka).
Invazivní houbové infekce
U pacientů léčených přípravkem Remicade by mělo být podezření na invazivní houbovou infekci, jako je aspergilóza, kandidóza, pneumocystóza, histoplazmóza, kokcidioidomykóza nebo blastomykóza, pokud se u nich rozvine závažné systémové onemocnění, a v rané fázi by měl být konzultován lékař kompetentní v diagnostice a léčbě invazivních mykotických infekcí při návštěvě těchto pacientů. Invazivní houbové infekce se mohou projevovat spíše jako diseminované než lokalizované onemocnění a testy antigenu a protilátek mohou být u některých pacientů s aktivní infekcí negativní. V diagnostickém procesu by měla být zvážena vhodná empirická antifungální terapie s přihlédnutím jak k riziku závažné houbové infekce, tak k rizikům antifungální terapie.
U pacientů, kteří žili nebo cestovali do oblastí, kde jsou endemické invazivní houbové infekce, jako je histoplazmóza, kokcidioidomykóza nebo blastomykóza, je třeba před zahájením léčby přípravkem Remicade pečlivě zvážit přínosy a rizika léčby přípravkem Remicade.
Fistulizující Crohnova choroba
Pacienti s fistulizující Crohnovou chorobou s akutními hnisavými píštělemi by neměli zahájit léčbu přípravkem Remicade, dokud nebude vyloučen zdroj možné infekce, zejména abscesů (viz bod 4.3).
Reaktivace hepatitidy B (HBV)
Reaktivace hepatitidy B byla pozorována u pacientů léčených antagonisty TNF, včetně infliximabu a kteří byli chronickými nositeli tohoto viru. V některých případech došlo k fatálním následkům.
Před zahájením léčby přípravkem Remicade by měli být pacienti vyšetřeni na infekci HBV.U pacientů, kteří mají pozitivní test na infekci HBV, se doporučuje konzultace s lékařem, který má zkušenosti s léčbou hepatitidy B.
Nosiče HBV vyžadující léčbu přípravkem Remicade by měly být pečlivě sledovány z hlediska známek a příznaků aktivní infekce HBV po celou dobu léčby a několik měsíců po jejím ukončení.Není k dispozici dostatek údajů o pacientech s HBV léčených antivirovou terapií v kombinaci s antagonisty TNF terapie k prevenci reaktivace HBV U pacientů, u kterých dojde k reaktivaci HBV, by měla být léčba Remicade přerušena a měla by být zahájena účinná antivirová terapie s vhodnou podpůrnou léčbou.
Hepatobiliární příhody
Během marketingového období Remicade byly pozorovány velmi vzácné případy žloutenky a neinfekční hepatitidy, některé s rysy autoimunitní hepatitidy. Byly ojedinělé případy selhání jater, které mělo za následek transplantaci jater nebo smrt. U pacientů se známkami a příznaky jaterní dysfunkce by měla být posouzena úroveň poškození jater. Pokud se vyvine žloutenka a / nebo zvýšení ALT ≥ 5násobek horní hranice normálu, léčba přípravkem Remicade by měla být ukončena a mělo by být provedeno důkladné vyšetření abnormálních stavů.
Asociace inhibitoru TNF-alfa a anakinry
V kombinovaných klinických studiích s anakinrou a jiným inhibitorem TNFα došlo k závažným infekcím a neutropenii, bez dalšího klinického prospěchu oproti použití samotného etanerceptu. Vzhledem k povaze nežádoucích účinků pozorovaných při kombinaci etanerceptu a anakinry se podobné kombinace anakinry a dalších inhibitorů TNFα. Proto se kombinace Remicade a anakinry nedoporučuje.
Asociace inhibitoru TNF-alfa a abataceptu
V klinických studiích bylo kombinované použití antagonistů TNF a abataceptu spojeno se zvýšeným rizikem infekcí, včetně závažných infekcí, ve srovnání s antagonisty TNF používanými samostatně, bez zvýšení klinického přínosu. Remicade a abatacept se nedoporučuje.
Spojení s jinými biologickými terapiemi
O souběžném používání infliximabu s jinými biologickými terapiemi používanými k léčbě stejných stavů jako infliximab nejsou dostatečné informace.Souběžné použití infliximabu s těmito biologickými přípravky se nedoporučuje z důvodu možnosti zvýšeného rizika infekce a dalších potenciálních lékových interakcí.
Substituce mezi biologickými DMARD
Při přechodu z jedné biologické látky na druhou je nutná opatrnost a pacienti by měli být nadále sledováni, protože překrývající se biologická aktivita může dále zvýšit riziko nežádoucích účinků, včetně infekce.
Živé vakcíny / infekční terapeutická činidla
U pacientů léčených anti-TNF terapií jsou k dispozici omezené údaje o odpovědi na očkování živými vakcínami nebo o sekundárním přenosu infekce při podání živých vakcín.Použití živých vakcín může vést ke klinickým infekcím, včetně diseminovaných infekcí. . Souběžné podávání živých vakcín s přípravkem Remicade se nedoporučuje.
U kojenců exponovaných in utero infliximabu byl po podání BCG vakcíny po narození hlášen smrtelný následek v důsledku diseminované infekce Calmette-Guérin bacillus (BCG). Před podáním živých vakcín exponovaným kojencům in utero pro infliximab se doporučuje čekací doba nejméně šest měsíců po porodu (viz bod 4.6).
Jiné použití infekčních terapeutických činidel, jako jsou živé oslabené bakterie (například intravezikální instilace pomocí BCG k léčbě rakoviny), by mohlo vést ke klinickým infekcím, včetně diseminovaných infekcí. Doporučuje se, aby terapeutická infekční agens nebyla podávána současně s Remicade.
Autoimunitní reakce
Relativní nedostatek TNFα způsobený terapií anti-TNF může vést k zahájení autoimunitního procesu.Pokud má pacient po léčbě přípravkem Remicade příznaky predikující syndrom podobný lupusu a je pozitivní na protilátky proti DNA na dvojitou šroubovici, ne měla by být zahájena další léčba přípravkem Remicade (viz bod 4.8).
Účinky na nervový systém
Použití látek blokujících TNF, včetně infliximabu, bylo spojeno s nástupem nebo exacerbací klinických příznaků a / nebo radiografických důkazů demyelinizačních poruch centrálního nervového systému, včetně roztroušené sklerózy, a periferních demyelinizačních poruch, včetně Guillain-Barrého syndromu U pacientů s před již existujícími nebo nedávnými demyelinizačními poruchami je třeba před zahájením léčby přípravkem Remicade pečlivě zvážit přínosy a rizika léčby anti-TNF.
Pokud se tyto stavy vyvinou, je třeba zvážit přerušení léčby přípravkem Remicade.
Zhoubné novotvary a lymfoproliferativní onemocnění
V kontrolovaných fázích klinických studií s inhibitory TNF bylo u pacientů, kteří dostali inhibitor TNF, pozorováno více případů malignit včetně lymfomu než u kontrolních pacientů. Během klinických studií s přípravkem Remicade byl ve všech schválených indikacích výskyt lymfomu u pacientů léčených přípravkem Remicade vyšší, než se očekávalo v obecné populaci, ale frekvence lymfomu byla vzácná. V postmarketingových zkušenostech byly hlášeny případy leukémie pacientů léčených antagonistou TNF. U pacientů s revmatoidní artritidou s velmi aktivním a dlouhodobým zánětlivým onemocněním, které komplikuje posouzení rizika, existuje zvýšené riziko vzniku lymfomu a leukémie.
V průzkumné klinické studii hodnotící použití přípravku Remicade u pacientů se středně těžkou až těžkou chronickou obstrukční plicní nemocí (Chronická obstrukční plicní nemoc(CHOPN), více případů maligních onemocnění bylo hlášeno u pacientů léčených přípravkem Remicade než u kontrolních pacientů. Všichni pacienti byli silní kuřáci. Je třeba věnovat pozornost hodnocení léčby pacientů se zvýšeným rizikem malignity jako silných kuřáků.
Na základě současných znalostí nelze vyloučit riziko vzniku lymfomů nebo malignit u pacientů léčených inhibitorem TNF (viz bod 4.8). Při zvažování léčby inhibitorem TNF u pacientů s anamnézou malignity nebo při zvažování prodloužení léčby u pacientů, u nichž se malignita rozvine, je třeba postupovat opatrně.
Opatrnosti je třeba také u pacientů s psoriázou, kteří byli v minulosti rozsáhle léčeni imunosupresivy nebo dlouhodobě PUVA.
Po uvedení přípravku na trh byly u dětí, dospívajících a mladých dospělých (do 22 let) léčených léky blokujícími TNF (zahájení terapie ≤ 18 let), včetně přípravku Remicade, hlášeny malignity, z nichž některé byly smrtelné. polovina případů byla lymfomy, ostatní případy zahrnovaly různé malignity a zahrnovaly vzácné malignity obvykle spojené s imunosupresí. Nelze vyloučit riziko vzniku maligních novotvarů u pacientů léčených inhibitory TNF.
Po uvedení přípravku na trh byly u pacientů léčených přípravky blokujícími TNF, včetně infliximabu, hlášeny vzácné případy hepatosplenického T-buněčného lymfomu (HSTCL). Tato vzácná forma T-buněčného lymfomu má extrémně agresivní průběh a výsledek. Obvykle smrtelný. pacienti byli léčeni AZA nebo 6-MP souběžně nebo bezprostředně před blokátorem TNF. Převážná většina případů s přípravkem Remicade se vyskytla u pacientů s Crohnovou chorobou nebo ulcerózní kolitidou a většina případů byla hlášena u dospívajících nebo mladých dospělých mužů. potenciální riziko kombinace AZA nebo 6-MP a Remicade musí být pečlivě zváženo. U pacientů léčených přípravkem Remicade nelze vyloučit riziko vzniku hepatosplenického T-buněčného lymfomu (viz bod 4.8).
U pacientů léčených blokátorem TNF včetně přípravku Remicade byl hlášen melanom a karcinom z Merkelových buněk (viz bod 4.8). Doporučuje se pravidelné kožní vyšetření, zejména u pacientů s rizikovými faktory rakoviny kůže.
Retrospektivní kohortová studie založená na datech ze švédských národních zdravotních rejstříků zjistila zvýšený výskyt rakoviny děložního čípku u žen s revmatoidní artritidou léčených infliximabem ve srovnání s biologicky neléčenými pacientkami nebo běžnou populací, včetně osob starších 60 let. U žen by měl pokračovat pravidelný screening léčeni přípravkem Remicade, včetně osob starších 60 let.
Všichni pacienti s ulcerózní kolitidou, u nichž je zvýšené riziko rozvoje dysplázie nebo karcinomu tlustého střeva (například pacienti s dlouhodobou ulcerózní kolitidou nebo primární sklerotizující cholangitidou) nebo kteří mají v anamnéze dysplázii nebo rakovinu tlustého střeva, by měli být vyšetřeni. k této dysplázii v pravidelných intervalech, před zahájením terapie a v průběhu onemocnění. Toto hodnocení by mělo zahrnovat kolonoskopii a biopsie podle místních pokynů.Na základě současných údajů není známo, zda léčba infliximabem ovlivňuje riziko vzniku dysplazie nebo rakoviny tlustého střeva (viz bod 4.8).
Protože možnost zvýšeného rizika vzniku rakoviny u pacientů léčených přípravkem Remicade s nově diagnostikovanou dysplazií nebyla stanovena, je nutné vyhodnotit poměr přínosů a rizik u jednotlivých pacientů a zvážit ukončení léčby.
Srdeční selhání
Remicade by měl být používán s opatrností u pacientů s mírným srdečním selháním (třída NYHA I / II). Pacienti by měli být pečlivě sledováni a léčba přípravkem Remicade by měla být ukončena u pacientů s novými nebo zhoršujícími se příznaky srdečního selhání (viz body 4.3 a 4.8).
Hematologické reakce
U pacientů užívajících anti-TNF léky, včetně přípravku Remicade, byly hlášeny případy pancytopenie, leukopenie, neutropenie a trombocytopenie. Všichni pacienti by měli být poučeni, aby okamžitě vyhledali lékařskou pomoc, pokud se u nich objeví kompatibilní příznaky nebo příznaky krevní dyskrazie (např. Přetrvávající horečka, podlitiny, krvácení a bledost). U pacientů s potvrzenými významnými hematologickými abnormalitami je třeba zvážit ukončení léčby přípravkem Remicade.
Ostatní
Zkušenosti s bezpečností léčby Remicade u pacientů, kteří podstoupili operaci, včetně artroplastiky, jsou omezené. Při plánování operace je třeba vzít v úvahu dlouhý eliminační poločas infliximabu. Pacienta, který během léčby přípravkem Remicade vyžaduje chirurgický zákrok, je třeba pečlivě sledovat, aby se zvýšilo riziko infekcí, a měla by být zvážena vhodná opatření.
Nereagování na léčbu Crohnovy choroby může znamenat přítomnost rigidních fibrotických striktur, které mohou vyžadovat chirurgickou léčbu. Neexistují žádné klinické důkazy, které by naznačovaly, že infliximab zhoršuje nebo způsobuje fibrotické striktury.
Zvláštní populace
Starší pacienti (≥ 65 let)
Výskyt závažných infekcí u pacientů ve věku 65 let a starších léčených přípravkem Remicade byl vyšší než u pacientů mladších 65 let. Některé z nich byly smrtelné. Při léčbě starších osob je třeba věnovat zvláštní pozornost riziku infekce (viz bod 4.8) .
Pediatrická populace
Infekce
V klinických studiích byly infekce hlášeny častěji u pediatrických pacientů než u dospělých (viz bod 4.8).
Očkování
Před zahájením léčby přípravkem Remicade se doporučuje pediatrickým pacientům, pokud je to možné, nechat se očkovat v souladu s nejnovějšími směrnicemi.
Zhoubné novotvary a lymfoproliferativní poruchy
Po uvedení přípravku na trh byly u dětí, dospívajících a mladých dospělých (do 22 let) léčených léky blokujícími TNF (zahájení terapie ≤ 18 let), včetně přípravku Remicade, hlášeny malignity, z nichž některé byly smrtelné. polovina případů byla lymfomy, ostatní případy zahrnovaly různé malignity a zahrnovaly vzácné malignity obvykle spojené s imunosupresí. Nelze vyloučit riziko vzniku malignit u dětí a dospívajících léčených inhibitory TNF.
Po uvedení na trh byly u pacientů léčených přípravky blokujícími TNF, včetně infliximabu, hlášeny vzácné případy hepatosplenického T-buněčného lymfomu. Tato vzácná forma T-buněčného lymfomu má extrémně agresivní průběh a obvykle fatální průběh. Téměř všichni pacienti byli léčeni s AZA nebo 6-MP souběžně nebo bezprostředně před blokátorem TNF. Převážná většina případů s přípravkem Remicade se vyskytla u pacientů s Crohnovou chorobou nebo ulcerózní kolitidou a většina případů byla hlášena u dospívajících nebo mladých dospělých mužů. Potenciální riziko kombinaci AZA nebo 6-MP a Remicade je třeba pečlivě zvážit. U pacientů léčených přípravkem Remicade nelze vyloučit riziko vzniku hepatosplenického T-buněčného lymfomu (viz bod 4.8).
04.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné interakční studie.
Existují náznaky, že současné užívání methotrexátu a jiných imunomodulátorů u pacientů s revmatoidní artritidou, psoriatickou artritidou a Crohnovou chorobou snižuje tvorbu protilátek proti infliximabu a zvyšuje plazmatické koncentrace infliximabu. Výsledky jsou však nejisté vzhledem k omezením metod použitých pro stanovení infliximabu a protilátek proti infliximabu v séru.
Kortikosteroidy zřejmě nemění farmakokinetiku infliximabu klinicky relevantním způsobem.
Kombinace přípravku Remicade s jinými biologickými terapiemi používanými k léčbě stejných stavů jako Remicade, včetně anakinry a abataceptu, se nedoporučuje (viz bod 4.4).
Doporučuje se nepodávat živé vakcíny současně s přípravkem Remicade. Doporučuje se také, aby kojenci po expozici nepodávali živé vakcíny in utero na infliximab po dobu nejméně 6 měsíců po porodu (viz bod 4.4).
Infekční terapeutická činidla by neměla být podávána současně s přípravkem Remicade (viz bod 4.4).
04.6 Těhotenství a kojení
Ženy v plodném věku
Ženy ve fertilním věku by měly během léčby přípravkem Remicade používat vhodnou antikoncepci a pokračovat v jejím užívání ještě nejméně 6 měsíců po poslední dávce.
Těhotenství
Mírný počet prospektivně shromážděných údajů o těhotných pacientkách (přibližně 450) léčených infliximabem se známými výsledky, včetně omezeného počtu (přibližně 230) těhotenství léčených během prvního trimestru, nevykazoval žádné neočekávané účinky na výsledek. Vzhledem k inhibici TNFα, infliximab podávaný během těhotenství může změnit normální imunitní reakce novorozence. Ve studii vývojové toxicity u myší s použitím podobné protilátky, která selektivně inhibuje funkčnost TNFα, nebyla zjištěna mateřská toxicita, embryotoxicita ani teratogenita (viz bod 5.3).
Dostupné klinické zkušenosti jsou příliš omezené na vyloučení rizik, a proto se podávání infliximabu během těhotenství nedoporučuje.
Infliximab prochází placentou a byl detekován v séru kojenců až 6 měsíců po narození. Po expozici in utero na infliximab mohou mít kojenci vyšší riziko infekce, včetně „závažné diseminované infekce, která může mít fatální následky. Podávání živých vakcín (např. BCG vakcína) exponovaným kojencům in utero infliximab se nedoporučuje podávat alespoň 6 měsíců po porodu (viz body 4.4 a 4.5). Byly také hlášeny případy agranulocytózy (viz bod 4.8).
Čas krmení
Není známo, zda se infliximab vylučuje do mateřského mléka nebo se po požití absorbuje systémově. Protože lidské imunoglobuliny jsou vylučovány do mateřského mléka, ženy by neměly kojit alespoň 6 měsíců po léčbě přípravkem Remicade.
Plodnost
K vyvození závěrů o účincích infliximabu na fertilitu a celkovou reprodukční funkci je k dispozici nedostatečné preklinické údaje (viz bod 5.3).
04.7 Účinky na schopnost řídit a obsluhovat stroje
Remicade má malý vliv na schopnost řídit nebo obsluhovat stroje. Po podání přípravku Remicade se může objevit závratě (viz bod 4.8).
04.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Infekce horních cest dýchacích byla nejběžnější nežádoucí reakcí (ADR) hlášenou v klinických studiích, vyskytující se u 25,3% pacientů léčených infliximabem oproti 16,5% u kontrolních pacientů. "Použití inhibitorů TNF hlášených u přípravku Remicade zahrnuje reaktivaci HBV, městnavé srdeční selhání ( CHF), závažné infekce (včetně sepse, oportunních infekcí a TBC), sérová nemoc (opožděné reakce z přecitlivělosti), hematologické reakce, systémový erytematosus / syndrom podobný lupusu, demyelinizační onemocnění, hepatobiliární příhody, lymfom, HSTCL, leukémie, karcinom z Merkelových buněk , melanom, dětská malignita, sarkoidóza / reakce sarkoidového typu, střevní nebo perianální absces (u Crohnovy choroby) a závažné reakce na infuzi (viz bod 4.4).
Tabulka nežádoucích účinků
Tabulka 1 uvádí seznam nežádoucích účinků hlášených v klinických studiích a nežádoucí účinky, některé s fatálním koncem, hlášené po uvedení na trh. V rámci třídy orgánových systémů jsou nežádoucí účinky seřazeny podle četnosti pomocí následujících kategorií: velmi časté (≥ 1/10); časté (≥ 1/100 až
stůl 1
Nežádoucí účinky v klinických studiích a po uvedení na trh
* včetně tuberkulózy skotu (diseminovaná BCG infekce), viz bod 4.4
Reakce související s infuzí
V klinických studiích byla reakce související s infuzí definována jako jakákoli nežádoucí událost, ke které došlo během infuze nebo do 1 hodiny po infuzi.V klinických studiích fáze III mělo 18% pacientů léčených infliximabem ve srovnání s 5% pacientů léčených placebem reakce související s infuzí. Celkově vyšší podíl pacientů, kteří dostali monoterapii infliximabem, zaznamenal reakci související s infuzí než pacienti, kteří dostávali souběžně infliximab s imunomodulátory.Přibližně 3% pacientů přerušilo léčbu v důsledku reakcí souvisejících s infuzí a všichni pacienti se zotavili s lékařskou terapií nebo bez ní.
Z pacientů léčených infliximabem, kteří měli reakci na infuzi během indukčního období do 6. týdne, 27% zažilo infuzní reakci během udržovacího období mezi 7. týdnem a 54. týdnem. U pacientů, kteří během indukčního období nereagovali na infuzi, 9% zaznamenalo reakci na infuzi během udržovacího období.
V klinické studii u pacientů s revmatoidní artritidou (ASPIRE) byly první 3 infúze podávány infuze po dobu 2 hodin. Trvání dalších infuzí bylo možné zkrátit na ne méně než 40 minut u pacientů, u kterých se neobjevily žádné reakce. infuzi. V této studii dostalo šedesát šest procent pacientů (686 z 1040) alespoň jednu zkrácenou infuzi trvající 90 minut nebo méně a 44% pacientů (454 z 1040) dostalo alespoň jednu zkrácenou infuzi trvající 60 minut nebo méně. U pacientů léčených infliximabem, kteří dostali alespoň jednu zkrácenou infuzi, se reakce související s infuzí objevily u 15% pacientů a závažné reakce s infuzí se vyskytly u 0,4% pacientů.
V klinické studii u pacientů s Crohnovou chorobou (SONIC) byly reakce související s infuzí pozorovány u 16,6% (27/163) pacientů léčených infliximabem v monoterapii, u 5% (9/179) pacientů léčených infliximabem v monoterapii. v kombinaci s AZA a u 5,6% (9/161) pacientů léčených monoterapií AZA. Závažná reakce na infuzi (
V období po uvedení přípravku na trh byly s podáním přípravku Remicade spojeny případy anafylaktoidních reakcí, včetně laryngeálního / faryngálního edému, závažného bronchospasmu a záchvatů.
Kromě toho byly také vzácně hlášeny přechodné ztráty zraku a ischemie myokardu / infarkt myokardu, ke kterým došlo během infuze přípravku Remicade nebo během dvou hodin (viz bod 4.4).
Reakce na infuzi po opakovaném podání přípravku Remicade
Klinická studie byla navržena u pacientů se středně těžkou až těžkou psoriázou za účelem zhodnocení účinnosti a bezpečnosti dlouhodobé udržovací terapie ve srovnání s opakovanou léčbou indukčním režimem Remicade (maximálně čtyři infuze v 0, 2, 6 a 14 týdnech) po onemocnění zhoršení. Pacienti nedostali žádnou souběžnou imunosupresivní terapii. V rameni s opakovanou léčbou došlo u 4% (8/219) pacientů k závažným infuzním reakcím na otok obličeje a hypotenzi. Ve všech případech byla léčba Remicade ukončena a / nebo byla přijata jiná léčba s úplným vymizením známek a symptomů.
Opožděná přecitlivělost
V klinických studiích byly opožděné reakce přecitlivělosti méně časté a vyskytovaly se po časových intervalech bez přípravku Remicade kratších než 1 rok. Ve studiích s psoriázou se opožděné reakce přecitlivělosti objevily na začátku léčby. Příznaky a příznaky zahrnovaly myalgii a / nebo artralgii s horečkou a / nebo vyrážkou, u některých pacientů svědění, otok obličeje, rukou nebo rtů, dysfagie, kopřivka, bolest v krku a bolest hlavy.
Není k dispozici dostatek údajů o výskytu opožděných reakcí z přecitlivělosti po časových intervalech bez přípravku Remicade delších než 1 rok, ale omezené údaje z klinických studií naznačují zvýšené riziko opožděné přecitlivělosti na prodloužení. Trvání časových intervalů bez podání přípravku Remicade (viz bod 4.4).
V roční klinické studii s opakovanými infuzemi u pacientů s Crohnovou chorobou (studie ACCENT I) byl výskyt reakcí vyplývajících z vývoje reakcí podobných sérovou nemocí 2,4%.
Imunogenita
U pacientů, u nichž se vyvinuly protilátky proti infliximabu, byla větší pravděpodobnost výskytu reakcí souvisejících s infuzí (přibližně 2 až 3krát vyšší).
V klinických studiích, ve kterých byly podávány jednotlivé a více dávek infliximabu v rozmezí od 1 do 20 mg / kg, byly protilátky proti infliximabu nalezeny u 14% pacientů léčených jakoukoli imunosupresivní terapií a u 24% pacientů bez imunosupresivní terapie. U 8% pacientů s revmatoidní artritidou opakovaně léčených doporučeným dávkováním a methotrexátem se vyvinuly protilátky proti infliximabu. U pacientů s psoriatickou artritidou léčených dávkou 5 mg / kg s nebo bez methotrexátu se protilátky vyvinuly celkem u 15% pacientů (u 4% pacientů užívajících methotrexát a 26% pacientů, kteří na začátku neužívali methotrexát) .U pacientů s Crohnovou chorobou, kteří dostávali udržovací léčbu, se vyvinuly protilátky proti infliximabu v průměru 3,3% pacientů užívajících imunosupresiva a 13,3% pacientů, kteří imunosupresiva nedostávali. krát vyšší u pacientů léčených epizodicky. Vzhledem k metodickým omezením negativní test nevyloučil přítomnost protilátek proti infliximabu. Někteří pacienti, u kterých se vyvinuly vysoké titry protilátek proti infliximabu, měli sníženou účinnost. U pacientů s psoriázou léčených udržovacím režimem infliximabu se při absenci souběžné léčby imunomodulátory vytvořily protilátky proti infliximabu přibližně 28% (viz bod 4.4: „Infuzní reakce a přecitlivělost“).
Infekce
U pacientů léčených přípravkem Remicade byla pozorována tuberkulóza, bakteriální infekce, včetně sepse a pneumonie, invazivní houbové, virové a jiné oportunní infekce. Některé z nich byly smrtelné. Mezi nejčastěji hlášené oportunní infekce s úmrtností> 5% patří pneumocystóza, kandidóza, listerióza a aspergilóza (viz bod 4.4).
V klinických studiích bylo 36% pacientů léčených infliximabem léčeno na infekce, ve srovnání s 25% pacientů léčených placebem.
V klinických studiích revmatoidní artritidy byl výskyt závažných infekcí včetně pneumonie vyšší u pacientů léčených infliximabem a methotrexátem než u pacientů léčených samotným methotrexátem, zejména v dávkách 6 mg / kg nebo vyšších (viz bod 4.4).
Mezi spontánními hlášeními hlášenými v období po uvedení přípravku na trh jsou nejčastějšími závažnými nežádoucími účinky infekce. Některé případy měly fatální následky. Téměř 50% hlášených úmrtí bylo spojeno s infekcí. Byly hlášeny případy tuberkulózy., někdy smrtelné, včetně případů miliární tuberkulózy a mimoplicní lokalizační tuberkulózy (viz bod 4.4).
Zhoubné novotvary a lymfoproliferativní onemocnění
V klinických studiích prováděných s infliximabem, ve kterých bylo léčeno 5 780 pacientů, což představuje 5 494 pacientoroků, bylo zjištěno 5 případů lymfomů a 26 případů maligních onemocnění bez lymfomu ve srovnání s žádnými případy lymfomu a 1 případem maligního onemocnění ne-lymfomu. pozorováno u 1600 pacientů léčených placebem, což představuje 941 pacientoroků.
V dlouhodobých studiích bezpečnosti s infliximabem po dobu až 5 let, což představuje 6 234 pacientoroků (3 210 pacientů), bylo hlášeno 5 případů lymfomu a 38 případů maligních onemocnění bez lymfomu.
V období po uvedení přípravku na trh byly také hlášeny případy malignit, včetně lymfomu (viz bod 4.4).
V průzkumné klinické studii zahrnující pacienty se středně těžkou až těžkou CHOPN, kteří byli kuřáci nebo bývalí kuřáci, bylo 157 dospělých pacientů léčeno přípravkem Remicade v podobných dávkách, jaké se používají při revmatoidní artritidě a Crohnově chorobě. U devíti z těchto pacientů se objevily malignity, včetně 1 lymfomu. Medián doby sledování byl 0,8 roku (incidence 5,7% [95% CI 2,65%-10,6%]. Mezi 77 pacienty v kontrolní skupině byl hlášen jeden případ malignity (medián doby sledování 0,8 roku; incidence 1,3% [95% CI 0,03% - 7,0%]). Většina těchto malignit zahrnovala plíce, hlavu nebo krk.
Populační retrospektivní kohortová studie zjistila zvýšený výskyt rakoviny děložního čípku u žen léčených infliximabem s revmatoidní artritidou ve srovnání s biologicky neléčenými pacienty nebo běžnou populací, včetně pacientů starších 60 let (viz bod 4.4).
Kromě toho byly po uvedení na trh u pacientů léčených přípravkem Remicade hlášeny vzácné případy hepatosplenického T-buněčného lymfomu, drtivá většina případů se vyskytla u pacientů s Crohnovou chorobou a ulcerózní kolitidou, většina pacientů byli dospívající nebo mladí dospělí muži (viz bod 4.4 ).
Srdeční selhání
Ve studii fáze II zaměřené na hodnocení přípravku Remicade u městnavého srdečního selhání (CHF) byl u pacientů léčených přípravkem Remicade, zejména u pacientů s nejvyšší dávkou 10 mg / kg, zjištěn vyšší výskyt úmrtnosti na zhoršení srdečního selhání. (tj. zdvojnásobit maximální schválenou dávku). V této studii bylo 150 pacientů s NYHA třídy III a IV CHF (ejekční frakce levé komory ≤ 35%) léčeno 3 infuzí přípravku Remicade 5 mg / kg, 10 mg / kg nebo placeba po dobu 6 týdnů. V 38. týdnu zemřelo 9 ze 101 pacientů léčených přípravkem Remicade (2 až 5 mg / kg a 7 až 10 mg / kg), zatímco mezi 49 pacienty léčenými placebem došlo k jednomu úmrtí.
V období po uvedení přípravku na trh byly u pacientů léčených přípravkem Remicade hlášeny případy zhoršení srdečního selhání, s identifikovatelnými spouštěči i bez nich. V období po uvedení přípravku na trh bylo také hlášeno nově vzniklé srdeční selhání, včetně srdečního selhání. žádné známé již existující kardiovaskulární onemocnění Někteří z těchto pacientů byli mladší 50 let.
Hepatobiliární příhody
V klinických studiích bylo u pacientů léčených přípravkem Remicade pozorováno mírné nebo střední zvýšení ALT a AST, aniž by došlo k závažnému poškození jater. Bylo pozorováno zvýšení ALT ≥ 5 x nad normální limity (ULN) (viz tabulka 2). Zvýšení aminotransferáz (častější u ALT než AST) bylo pozorováno u většího podílu pacientů léčených přípravkem Remicade než u kontrolních skupin, a to jak při podávání přípravku Remicade samostatně, tak v kombinaci s jinými imunosupresivy. Většina abnormalit aminotransferáz byla přechodná; u malého počtu pacientů však došlo k prodlouženému nárůstu. Obecně byli pacienti, u kterých došlo ke zvýšení hladin ALT a AST, asymptomatičtí a abnormality se snížily nebo odezněly buď pokračováním nebo ukončením léčby přípravkem Remicade nebo změnou souběžné léčby. U pacientů užívajících přípravek Remicade byly v období postmarketingového sledování hlášeny velmi vzácné případy žloutenky a hepatitidy, některé s rysy autoimunitní hepatitidy (viz bod 4.4).
Tabulka 2
Počet pacientů se zvýšenou aktivitou ALT v klinických studiích
1 Pacienti ve skupině s placebem dostávali methotrexát, zatímco pacienti ve skupině s infliximabem dostávali infliximab i methotrexát.
2 Pacienti ve skupině s placebem ve 2 studiích fáze III Crohnovy choroby, ACCENT I a ACCENT II, dostali úvodní dávku 5 mg / kg infliximabu při zahájení studie a placebo v udržovací fázi. Byli randomizováni do udržovací skupiny s placebem. a následně přešli na infliximab, byli zařazeni do skupiny infliximabu do ALT analýzy. Ve studii fáze IIIb u Crohnovy choroby, SONIC, dostávali pacienti v rameni s placebem AZA 2,5 mg / kg / den jako aktivní kontrolu, navíc k infuzi placeba s infliximabem.
3 Počet pacientů hodnocených na ALT.
4 Průměrné sledování je založeno na léčených pacientech.
Antinukleární protilátky (ANA) / dvouřetězcové DNA protilátky (dsDNA)
Asi polovina pacientů léčených infliximabem v klinických studiích, kteří byli na počátku ANA negativní, se během studie stala ANA pozitivní, ve srovnání s přibližně jednou pětinou pacientů léčených placebem. Protilátky anti-dsDNA byly nedávno detekovány přibližně u 17% pacientů léčených infliximabem ve srovnání s 0% pacientů léčených placebem. V nejnovějším hodnocení zůstalo 57% pacientů léčených infliximabem pozitivních na protilátky anti-dsDNA. Zprávy o podobném lupusu a lupusových syndromech však zůstávají vzácné (viz bod 4.4).
Pediatrická populace
Pacienti s juvenilní revmatoidní artritidou
Remicade byl studován v klinické studii zahrnující 120 pacientů (věkové rozmezí: 4–17 let) s aktivní juvenilní revmatoidní artritidou bez ohledu na použití methotrexátu. Pacienti byli léčeni infliximabem 3 nebo 6 mg / kg jako 3dávkový indukční režim (týden 0 (2, 6, respektive 14, 16, 20, v tomto pořadí) s následnou udržovací terapií každých 8 týdnů v kombinaci s methotrexátem.
Infuzní reakce
Infuzní reakce se vyskytly u 35% pacientů s juvenilní revmatoidní artritidou, kteří dostávali 3 mg / kg, ve srovnání se 17,5% pacientů, kteří dostávali 6 mg / kg. Ve skupině s přípravkem Remicade 3 mg / kg došlo u 4 ze 60 pacientů k závažné reakci na infuzi a u 3 pacientů možná anafylaktická reakce (2 z nich byly zahrnuty do závažných reakcí na infuzi). Ve skupině s dávkou 6 mg / kg došlo u 2 z 57 pacientů k závažné reakci na infuzi. “infuze, z nichž jeden měl možnou anafylaktickou reakci (viz bod 4.4 ).
Imunogenita
U 38% pacientů užívajících 3 mg / kg se vyvinuly protilátky proti infliximabu ve srovnání s 12% pacientů dostávajících 6 mg / kg. Titry protilátek byly významně vyšší ve skupině dostávající 3 mg / kg než ve skupině dostávající 6 mg / kg.
Infekce
Infekce se vyskytla u 68% (41/60) dětí, které dostaly 3 mg / kg po dobu 52 týdnů, u 65% (37/57) dětí, které dostaly 6 mg / kg infliximabu po dobu 38 týdnů, a u 47% (28 /60) dětí užívajících placebo po dobu 14 týdnů (viz bod 4.4).
Dětští pacienti s Crohnovou chorobou
Následující nežádoucí účinky byly hlášeny častěji u dětských pacientů s Crohnovou chorobou zahrnutých do studie REACH (viz bod 5.1) než u dospělých pacientů s Crohnovou chorobou: anémie (10,7%), krev ve stolici (9,7%), leukopenie (8,7%), zrudnutí se zarudnutím kůže (8,7%), virovými infekcemi (7,8%), neutropenií (6,8%), zlomeninami kostí (6,8%), bakteriálními infekcemi (5,8%) a alergickými reakcemi postihujícími dýchací cesty (5,8%). Další zvláštní aspekty jsou uvedeny níže.
Reakce související s infuzí
17,5% randomizovaných pacientů ve studii REACH zažilo 1 nebo více reakcí na infuzi.Nebyly hlášeny žádné závažné případy reakcí na infuzi a u 2 subjektů ve studii REACH se vyvinuly nezávažné anafylaktické reakce.
Imunogenita
Protilátky proti infliximabu byly detekovány u 3 (2,9%) pediatrických pacientů.
Infekce
Ve studii REACH byly infekce hlášeny u 56,3% randomizovaných subjektů léčených infliximabem. Infekce byly hlášeny častěji u subjektů, které dostávaly infuze každých 8 týdnů, než u pacientů léčených každých 12 týdnů (73,6%, respektive 38,0%), zatímco závažné infekce byly hlášeny u 3 subjektů ve skupinové udržovací léčbě každých 8 týdnů a u 4 subjektů v léčená skupina každých 12 týdnů. Nejčastěji hlášenými infekcemi byly infekce horních cest dýchacích a faryngitida. Nejčastější ze závažných infekcí byl absces. Byly hlášeny 3 případy zápalu plic (1 závažný) a 2 případy herpes zoster (oba nezávažné).
Dětští pacienti s ulcerózní kolitidou
Celkově byly nežádoucí účinky hlášené ve studii u pediatrických pacientů s ulcerózní kolitidou (C0168T72) obecně v souladu s těmi, které byly hlášeny ve studiích u dospělých pacientů s ulcerózní kolitidou (ACT 1 a ACT 2). Ve studii C0168T72 byly nejčastějšími nežádoucími účinky infekce horních cest dýchacích, faryngitida, bolest břicha, horečka a bolest hlavy. Nejčastějším nežádoucím účinkem bylo zhoršení ulcerózní kolitidy, jejíž výskyt byl vyšší u pacientů léčených každých 12 týdnů než u pacientů léčených každých 8 týdnů.
Reakce související s infuzí
Celkově 8 (13,3%) ze 60 léčených pacientů uvedlo jednu nebo více reakcí na infuzi, přičemž 4 z 22 pacientů (18,2%) v udržovací skupině každých 8 týdnů a 3 z 23 pacientů (13,0%) v léčené udržovací skupině každých 12 týdnů Nebyly hlášeny žádné závažné reakce na infuzi. Všechny reakce na infuzi byly mírné nebo střední intenzity.
Imunogenita
Protilátky proti infliximabu byly detekovány u 4 (7,7%) pacientů do 54. týdne.
Infekce
Infekce byly hlášeny u 31 (51,7%) ze 60 pacientů léčených ve studii C0168T72 a 22 (36,7%) vyžadovalo orální nebo parenterální antimikrobiální léčbu. Podíl pacientů s infekcemi ve studii C0168T72 byl podobný jako ve studii dětské Crohnovy choroby (REACH), ale vyšší než podíl ve studiích dospělé ulcerózní kolitidy (ACT 1 a ACT 2). Celkový výskyt infekcí ve studii C0168T72 byl 13/22 (59%) ve udržovací skupině léčené každých 8 týdnů a 14/23 (60,9%) ve udržovací skupině léčené každých 12 týdnů. Horní cesty dýchací (7/60 [12 %]) a faryngitida (5/60 [8%]) byly nejčastěji hlášenými infekcemi dýchacího systému. Závažné infekce byly hlášeny u 12% (7/60) všech léčených pacientů.
V této studii bylo více pacientů ve skupině 12 až 17 let než ve skupině 6 až 11 let (45/60 [75,0%]) vs. 15/60 [25,0%]).Ačkoli počet pacientů v každé podskupině byl příliš malý na to, aby bylo možné učinit nějaké pevné závěry ohledně „vlivu věku na události související s bezpečností“, byl vyšší podíl pacientů se závažnými nežádoucími účinky a přerušením léčby způsobeným událostmi. mladší věková skupina než ve starší věkové skupině. Přestože podíl pacientů s infekcemi byl také vyšší v mladší věkové skupině, u závažných infekcí byly podíly v těchto dvou skupinách podobné. Celkový podíl nežádoucích účinků a reakcí na infuzi byl podobné mezi věkovými skupinami 6 až 11 let a 12 až 17 let.
Postmarketingové zkušenosti
Spontánní postmarketingové hlášení závažných nežádoucích účinků u pediatrických pacientů zahrnovalo malignity včetně hepatosplenického T-buněčného lymfomu, přechodné abnormality jaterních enzymů, lupus-like syndromy a pozitivní na autoprotilátky (viz body 4.4 a 4.8).
Další informace o zvláštních populacích
Starší pacienti (≥ 65 let)
V klinických studiích s revmatoidní artritidou byl výskyt závažných infekcí vyšší u pacientů léčených infliximabem plus methotrexátem ve věku 65 a více let (11,3%) než u pacientů mladších 65 let (4, 6%). U pacientů léčených samotným methotrexátem byl výskyt závažných infekcí 5,2% u pacientů ve věku 65 let a starších ve srovnání s 2,7% u pacientů mladších 65 let (viz bod 4.4).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky, které se vyskytnou po registraci léčivého přípravku, je důležité, protože umožňuje průběžné sledování poměru přínosu a rizika léčivého přípravku. Zdravotničtí pracovníci jsou požádáni, aby hlásili jakékoli podezření na nežádoucí účinky na adresu Italská agentura pro léčivé přípravky. , webové stránky: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Předávkování
Nebyly hlášeny žádné případy předávkování. Jednotlivé dávky až do maximálně 20 mg / kg byly podávány bez toxických účinků.
05.0 FARMAKOLOGICKÉ VLASTNOSTI
05.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: inhibitory tumor nekrotizujícího faktoru alfa (TNF-alfa).
ATC kód: L04AB02.
Mechanismus účinku
Infliximab je chimérická lidská myší monoklonální protilátka, která se s vysokou afinitou váže na rozpustnou i transmembránovou formu TNFα, nikoli však na lymfotoxin α (TNFβ).
Farmakodynamické účinky
Infliximab inhibuje in vitro Aktivita TNFa v široké škále biologických dávek Infliximab zabraňoval onemocnění u transgenních myší, u kterých se vyvinula polyartritida jako důsledek esenciální exprese lidského TNFa, a když byl podáván po nástupu onemocnění, umožnil regresi kloubních erozí. In vivo, infliximab rychle vytváří stabilní komplexy s lidským TNFα, což je proces, který vede ke ztrátě biologické aktivity TNFα.
V kloubech pacientů s revmatoidní artritidou byly detekovány vysoké koncentrace TNFα a souvisí s vysokou aktivitou onemocnění.Léčba infliximabem vedla ke snížení infiltrace zánětlivých buněk v zanícených oblastech kloubů a ke snížení exprese při revmatoidní artritidě molekuly zprostředkující buněčnou adhezi, chemotaxi a degradaci tkáně. Po léčbě infliximabem došlo u pacientů ke snížení hladin interleukinu 6 (IL-6) a C-reaktivního proteinu (CRP) v séru a zvýšených hladin hemoglobinu u pacientů se sníženou hladinou hemoglobinu ve srovnání s hodnotami před léčbou. Lymfocyty periferní krve navíc nevykazovaly významný pokles počtu a proliferativní odpovědi na test in vitro mitogenní stimulace ve srovnání s buňkami neléčených pacientů. U pacientů s psoriázou vedla léčba infliximabem ke snížení epidermálního zánětu a normalizaci diferenciace keratinocytů na psoriatické plaky. U psoriatické artritidy snížila krátkodobá léčba přípravkem Remicade počet T buněk a cév. V synoviální a psoriatické kůži.
Histologické vyhodnocení biopsií tlustého střeva provedené před a 4 týdny po podání infliximabu odhalilo podstatné snížení detekovatelného TNFα. Léčba infliximabem u pacientů s Crohnovou chorobou byla také významně spojena
snížení sérové koncentrace CRP, běžně zvýšeného zánětlivého markeru. Celkový počet periferních leukocytů byl u pacientů léčených infliximabem ovlivněn minimálně, přestože změny lymfocytů, monocytů a neutrofilů odrážely změny od normálních hodnot. Mononukleární buňky periferní krve (PBMC) pacientů léčených infliximabem vykazovaly ve srovnání s neléčenými pacienty neovlivněnou schopnost proliferativní reakce na podněty; dále po ošetření infliximabem nebyly pozorovány žádné podstatné změny v produkci cytokinů stimulovanými buňkami PBMC. Analýza mononukleárních buněk lamina propria získaných po biopsii střevní sliznice ukázala, že léčba infliximabem vedla ke snížení počtu buněk schopných exprimovat TNFα a interferon γ. Další histologické studie poskytly důkaz, že léčba infliximabem snižuje infiltraci zánětlivých buněk do postižených oblastí střeva a přítomnost popisovačEndoskopické studie střevní sliznice prokázaly hojení sliznice u pacientů léčených infliximabem.
Klinická účinnost a bezpečnost
Artritida revmatoid u dospělých
Účinnost infliximabu byla hodnocena ve dvou multicentrických, randomizovaných, dvojitě zaslepených pilotních klinických studiích: ATTRACT a ASPIRE. V obou studiích souběžné užívání stabilních dávek kyseliny listové, perorálních kortikosteroidů (≤ 10 mg / die) a / nebo non- steroidní protizánětlivé léky (NSAID).
Primárními cílovými ukazateli byla redukce příznaků a symptomů, jak jsou definována kritérii American College of Rheumatology (ACR20 pro ATTRACT, indikátor ACR-N pro ASPIRE), prevence strukturálního poškození kloubů a zlepšení fyzických funkcí. A symptomy byly definovány jako zlepšení nejméně 20% (ACR20) v počtu bolestivých a oteklých kloubů a ve 3 z následujících 5 kritérií: celkové hodnocení lékaře, celkové hodnocení pacienta, hodnocení funkce / postižení, vizuální analogová stupnice bolesti, rychlost sedimentace erytrocytů nebo C- reaktivní protein. ACR-N používá stejná kritéria jako ACR20, vypočtená s ohledem na nejnižší procento zlepšení počtu oteklých kloubů, bolestivých kloubů a mediánu zbývajících 5 složek odpovědi ACR. Strukturální poškození kloubů (eroze a redukce kloubní linie) v rukou a nohou bylo měřeno vyhodnocením změny celkového Sharpova skóre modifikovaného van der Heijde (0-440) oproti výchozímu stavu. K posouzení průměrné změny fyzických funkcí v čase od výchozí hodnoty byl použit dotazník pro hodnocení zdraví (HAQ; škála 0 až 3).
Studie ATTRACT hodnotila odpovědi ve 30., 54. a 102. týdnu v placebem kontrolované studii u 428 pacientů s aktivní revmatoidní artritidou navzdory léčbě methotrexátem. Přibližně 50% pacientů bylo ve funkční třídě III. Pacienti byli léčeni placebem, 3 mg / kg nebo 10 mg / kg infliximabu v týdnech 0, 2 a 6 a poté každé 4 nebo 8 týdnů. Všichni pacienti užívali stabilní dávku methotrexátu (medián 15 mg / týden) po dobu 6 měsíců před zařazením a zůstali na stabilních dávkách po celou dobu studie.
Výsledky v 54. týdnu (ACR20, van der Heijde-modifikované celkové Sharp skóre a HAQ), jsou uvedeny v tabulce 3. Vyšší výskyt klinické odpovědi (ACR50 a ACR70) byl pozorován ve všech skupinách léčených infliximabem ve 30. a 54. týdnu ve srovnání se samotným methotrexátem.
Snížení rychlosti progrese strukturálního poškození kloubů (eroze a zmenšení mezery kloubu) bylo pozorováno ve všech skupinách léčených infliximabem po 54 týdnech (tabulka 3).
Účinky pozorované v 54. týdnu byly zachovány do 102. týdne léčby. Vzhledem k počtu přerušení léčby nebylo možné definovat rozsah účinného rozdílu mezi skupinami s monoterapií infliximabem a methotrexátem.
Tabulka 3
Účinky na ACR20, strukturální poškození kloubů a fyzické funkce v 54. týdnu, ATTRACT
kontrolované = Všichni pacienti měli aktivní RA navzdory léčbě stabilními dávkami methotrexátu po dobu 6 měsíců před zařazením do studie a během studie museli zůstat na stabilních dávkách.Souběžné užívání stabilních dávek perorálních kortikosteroidů (≤ 10 mg / den) a / nebo bylo povoleno nesteroidní protizánětlivé léčivo (NSAID); byl podán folátový doplněk.
b Všechny dávky infliximabu byly podávány současně s methotrexátem a folátem a v některých případech s kortikosteroidy a / nebo nesteroidními protizánětlivými léky (NSAID)
c p
d vyšší hodnoty znamenají větší poškození kloubů.
a HAQ = dotazník pro hodnocení zdraví; vyšší hodnoty znamenají „menší postižení“.
Studie ASPIRE hodnotila reakce v 54. týdnu u 1004 pacientů dosud neléčených methotrexátem s aktivní revmatoidní artritidou (medián počtu oteklých a citlivých kloubů: 19, respektive 31) s nedávným nástupem (trvání onemocnění ≤ 3 roky, medián 0,6 roku). Všichni pacienti dostávali methotrexát (optimalizovaný na 20 mg / týden do 8. týdne) v kombinaci s placebem nebo infliximabem 3 mg / kg nebo 6 mg / kg v týdnech 0, 2 a 6 a poté každých 8 týdnů. Výsledky v 54. týdnu jsou uvedeny v tabulce 4.
Po 54 týdnech léčby obě dávky infliximabu + methotrexátu poskytly statisticky významně větší zlepšení známek a symptomů než samotný methotrexát, měřeno podílem pacientů dosahujících odpovědi ACR 20, 50 a 70.
V ASPIRE mělo více než 90% pacientů alespoň dva vyhodnotitelné rentgenové snímky. Snížení rychlosti progrese strukturálního poškození bylo pozorováno ve 30. a 54. týdnu ve skupinách infliximab + methotrexát ve srovnání se samotným methotrexátem.
Tabulka 4
Účinky na ACRn, strukturální poškození kloubů a fyzické funkce v 54. týdnu, ASPIRE
na str
b vyšší hodnoty znamenají větší poškození kloubů.
c dotazník pro hodnocení zdraví; vyšší hodnoty znamenají „menší postižení“.
d p = 0,030 e
Údaje na podporu titrace dávky u revmatoidní artritidy pocházejí ze studií
Přitáhněte, aspirujte a začněte. START byla randomizovaná, multicentrická, dvojitě zaslepená, tříramenná, paralelní skupina, bezpečnostní studie. V jednom ze studijních ramen (skupina 2, n = 329) byla pacientům s nedostatečnou odpovědí povolena titrace dávky v přírůstcích po 1,5 mg / kg od 3 do 9 mg / kg. Většina (67%) těchto pacientů nevyžadovala titraci dávky. Z pacientů, kteří to vyžadovali, dosáhlo 80% klinické odpovědi a většina (64%) z nich vyžadovala pouze zvýšení o 1,5 mg / kg.
Crohnova choroba u dospělých
Indukční léčba aktivní, středně těžké až těžké Crohnovy choroby Účinnost jednorázové dávky infliximabu byla hodnocena u 108 pacientů s aktivní Crohnovou chorobou v rozsahu od středně těžkých až po závažné (index aktivity Crohnovy choroby (CDAI) ≥ 220 ≤ 400) ve studii randomizované, dvojitě zaslepené, placebem kontrolované dávky. odpověď. Ze 108 pacientů bylo 27 léčeno doporučenou dávkou infliximabu (5 mg / kg). Všichni pacienti měli nedostatečnou odpověď na předchozí konvenční terapie Souběžné použití nezměněných dávek konvenčních terapií bylo povoleno a 92% pacientů poté pokračovalo přijímat takové terapie.
Primárním cílovým parametrem byl výpočet počtu pacientů s klinickou odpovědí, definovanou jako pokles CDAI o ≥ 70 bodů oproti výchozím hodnotám, ve 4. týdnu a bez nárůstu užívání léků nebo chirurgických zákroků pro tuto nemoc. Crohn. Pacienti, kteří reagovali ve 4. týdnu, byli sledováni až do 12. týdne. Sekundární cílové parametry zahrnovaly počet pacientů v klinické remisi ve 4. týdnu (CDAI
Ve 4. týdnu po podání jednotlivé dávky mělo 22/27 (81%) pacientů léčených infliximabem v dávce 5 mg/kg klinickou odpověď ve srovnání se 4/25 (16%) pacienty léčenými placebem (p
Udržovací léčba u aktivní, středně těžké až těžké Crohnovy choroby u dospělých
Účinnost opakovaných infuzí s infliximabem byla hodnocena v 1leté klinické studii (ACCENT I). Celkem 573 pacientů se středně těžkou až těžkou aktivní Crohnovou chorobou (CDAI ≥ 220 ≤ 400) dostalo jednu infuzi 5 mg / kg týdně 0,178 z 580 zařazených pacientů (30,7%) mělo závažné onemocnění (skóre CDAI> 300 a souběžná léčba kortikosteroidy a / nebo imunosupresivy) odpovídající populaci definované v indikacích (viz bod 4.1) Ve 2. týdnu byli všichni pacienti hodnoceni pro klinickou odpověď a randomizováni do jedné ze 3 léčebných skupin; udržovací skupina s placebem, udržovací skupina 5 mg / kg a udržovací dávka 10 mg / kg. Poté všechny 3 skupiny dostaly opakované infuze ve 2., 6. týdnu a poté každých 8 týdnů .
Z 573 randomizovaných pacientů dosáhlo 335 (58%) klinické odpovědi ve 2. týdnu. Tito pacienti byli klasifikováni jako respondéři 2. týdne a zahrnuti do primární analýzy (viz tabulka 5). 2. týden, 32% (26/81) v skupina s udržováním placeba a 42% (68/163) ve skupině s udržovací infliximabem dosáhlo klinické odpovědi v 6. týdnu. Poté nebyl mezi skupinami žádný rozdíl v počtu pacientů, kteří následně reagovali na léčbu.
Koprimárními koncovými body bylo procento pacientů v klinické remisi (CDAI
Tabulka 5
Účinky na rychlost reakce a remise, údaje z ACCENT I (pacienti dosahující odpovědi ve 2. týdnu)
a Snížení CDAI o ≥ 25% a ≥ 70 bodů.
b CDAI
Na začátku 14. týdne byli pacienti, kteří reagovali na léčbu, ale následně ztratili klinický přínos, převedeni na dávku infliximabu o 5 mg / kg vyšší, než byla dávka, na kterou byli původně randomizováni. L "Osmdesát devět procent (50 / 56) pacientů, kteří ztratili klinickou odpověď na udržovací léčbu infliximabem 5 mg / kg po 14. týdnu, odpověděli na léčbu infliximabem 10 mg / kg.
Ve 30. a 54. týdnu bylo ve skupinách s udržovací léčbou infliximabem ve srovnání se skupinou s udržováním placeba pozorováno zlepšení v hodnocení kvality života, snížení hospitalizací souvisejících s onemocněním a používání kortikosteroidů.
Infliximab, s nebo bez AZA, byl hodnocen v randomizované, dvojitě zaslepené studii aktivního komparátoru (SONIC) s 508 dospělými pacienty se středně těžkou až těžkou Crohnovou chorobou (CDAI ≥ 220 ≤ 450), kteří nikdy předtím nebyli léčeni biologiemi a imunosupresiva a se střední dobou trvání onemocnění 2,3 roku. Na začátku bylo 27,4% pacientů na systémových kortikosteroidech, 14,2% pacientů na budesonidu a 54,3% pacientů na 5-ASA sloučeninách. Pacienti byli randomizováni k monoterapii AZA, monoterapii infliximabem nebo kombinované léčbě infliximab plus AZA. Infliximab byl podáván v dávce 5 mg / kg v týdnech 0, 2, 6 a poté každých 8 týdnů. AZA byla podávána v denní dávce 2,5 mg / kg.
Primárním cílovým parametrem studie byla klinická remise bez kortikosteroidů v 26. týdnu, definovaná jako pacienti s klinickou remisí (CDAI prednison nebo ekvivalent) nebo budesonid v dávce> 6 mg / den. Výsledky viz tabulka 6. Podíl pacientů s hojením sliznic v 26. týdnu byly významně vyšší ve skupinách s kombinací infliximab plus AZA (43,9%, s
Tabulka 6
Procento pacientů, kteří dosáhli klinické remise bez kortikosteroidů v 26. týdnu, SONIC
* P-hodnoty představují každou skupinu léčenou infliximabem oproti monoterapii AZA
Podobné vzorce v dosažení klinické remise bez kortikosteroidů byly pozorovány v 50. týdnu. Kromě toho bylo u infliximabu pozorováno zlepšení kvality života, jak uvádí dotazník IBDQ.
Indukční léčba aktivní fistulizující Crohnovy choroby
Účinnost byla hodnocena v randomizované, dvojitě zaslepené, placebem kontrolované studii u 94 pacientů s Crohnovou chorobou, kteří měli píštěle po dobu alespoň 3 měsíců. Jednačtyřicet z těchto pacientů bylo léčeno 5 mg / kg infliximabu. Asi 93% pacientů dříve podstoupilo antibiotickou nebo imunosupresivní terapii.
Bylo povoleno současné užívání konvenčních terapií v nezměněné dávce a 83% pacientů pokračovalo v léčbě alespoň jednou z těchto terapií.Pacienti dostali tři dávky placeba nebo infliximabu v týdnech 0, 2 a 6. Sledování pacientů pacientů bylo 26 týdnů . Primárním cílovým parametrem byl počet pacientů s klinickou odpovědí, definovanou jako ≥ 50% snížení počtu vyplachovacích píštělí oproti výchozím hodnotám po mírném stlačení alespoň ve dvou po sobě jdoucích kontrolách (o 4 týdny později), bez zvýšení užívání drog nebo chirurgický zákrok pro Crohnovu chorobu.
68% (21/31) pacientů, kterým byl podáván infliximab v dávce 5 mg/kg, zaznamenalo klinickou odpověď ve srovnání s 26% (8/31) pacientů léčených placebem (p = 0,002). Průměrná doba do odpovědi ve skupině s infliximabem byla 2 týdny. Průměrná doba odpovědi byla 12 týdnů. Kromě toho bylo u 55% pacientů léčených infliximabem pozorováno uzavření všech píštělí, ve srovnání s 13% pacientů užívajících placebo (p = 0,001).
Udržovací léčba u aktivní fistulizující Crohnovy choroby
Účinnost opakovaných infuzí infliximabu u pacientů s fistulizující Crohnovou chorobou byla hodnocena v roční studii (ACCENT II). Celkem 306 pacientů dostalo 3 dávky 5 mg / kg infliximabu v týdnech 0, 2 a 6. Na začátku , 87% pacientů mělo perianální píštěle, 14% mělo břišní píštěle, 9% mělo rektovaginální píštěl. Střední skóre CDAI bylo 180. Ve 14. týdnu bylo hodnoceno 282 pacientů z hlediska klinické odpovědi a randomizováno k léčbě placebem nebo infliximabem 5 mg / kg každých 8 týdnů až 46. týden.
Pacienti, kteří odpověděli ve 14. týdnu (195/282), byli analyzováni na primární cílový parametr, kterým byla doba mezi randomizací a ztrátou odpovědi (viz tabulka 7). Po 6. týdnu byly povoleny snížené kortikosteroidy.
Tabulka 7
Účinky na rychlost reakce, údaje ze studie ACCENT II (pacienti dosahující odpovědi ve 14. týdnu)
a ≥ 50% snížení počtu drénujících píštělí oproti výchozímu stavu po dobu ≥ 4 týdnů
b Absence drenážních píštělí
Na začátku 22. týdne byli pacienti, kteří zpočátku reagovali na léčbu a následně ztratili odpověď, převedeni na aktivní opakovanou léčbu každých 8 týdnů v dávce infliximabu o 5 mg / kg vyšší, než byla dávka, na kterou byli původně randomizováni. skupina udržující infliximab 5 mg / kg, která přešla na aktivní opětovné léčení, protože po 22. týdnu ztratila odpověď na snížení píštěle, 57% (12/21) reagovalo na opakovanou léčbu infliximabem 10 mg / kg každých 8 týdnů.
Nebyl žádný významný rozdíl mezi placebem a infliximabem v poměru pacientů s trvalým uzavřením všech píštělí do 54. týdne, v příznacích proktalgie, infekcí abscesu a močových cest nebo v počtu nových píštělí vyvinutých během léčby.
Udržovací terapie infliximabem každých 8 týdnů významně snížila hospitalizace a chirurgické zákroky ve srovnání s placebem. Kromě toho bylo pozorováno snížení používání kortikosteroidů a zlepšení kvality života.
Ulcerózní kolitida u dospělých
Bezpečnost a účinnost přípravku Remicade byla hodnocena ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných klinických studiích (ACT 1 a ACT 2) u dospělých pacientů se středně těžkou až těžkou aktivní ulcerózní kolitidou (Mayo skóre 6 až 12; endoskopické dílčí skóre ≥ 2) s neadekvátní odpovědí na konvenční terapie [perorální kortikosteroidy, aminosalicyláty a / nebo imunomodulátory (6 MP, AZA)]. Bylo povoleno současné podávání fixních dávek perorálních aminosalicylátů, kortikosteroidů a / nebo imunolodulačních léků. Pacienti ve studiích byli randomizováni k podávání placeba nebo Remicade 5 mg / kg nebo Remicade 10 mg / kg v týdnech 0, 2, 6, 14 a 22 a v ACT 1 v týdnech 30, 38 a 46. Redukce kortikosteroidů byla povolena po 8 týdnech.
Tabulka 8
Účinky na klinickou odpověď, klinickou remisi a slizniční hojení v 8. a 30. týdnu.
Kombinovaná data z ACT 1 a 2
na str
Účinnost přípravku Remicade v 54. týdnu byla hodnocena ve studii ACT 1.
V 54. týdnu měla klinickou odpověď 44,9% pacientů ve skupině s kombinací infliximabu ve srovnání s 19,8% ve skupině s placebem (p
Větší část pacientů ve skupině s kombinací infliximabu byla schopna přerušit léčbu kortikosteroidy a zůstat v klinické remisi ve srovnání se skupinou s placebem po oba 30. týdny (22,3% vs. 7,2%, p
Kombinovaná data ze studií ACT 1 a ACT 2 a jejich rozšíření analyzovaná od výchozích hodnot do 54. týdne prokázala snížení hospitalizací souvisejících s ulcerózní kolitidou a chirurgických zákroků po léčbě infliximabem. Počet hospitalizací souvisejících s ulcerózní kolitidou byl významně nižší v léčebných skupinách s infliximabem 5 a 10 mg / kg ve srovnání se skupinou s placebem (průměrný počet hospitalizací na 100 subjektů za rok: 21 a 19 oproti 40 ve skupině s placebem; p = 0,019, respektive p = 0,007).
Počet chirurgických zákroků souvisejících s ulcerózní kolitidou byl také nižší ve skupinách léčených infliximabem 5 a 10 mg / kg ve srovnání se skupinou s placebem (průměrný počet operací na 100 subjektů za rok: 22 a 19 oproti 34; p = 0,145 a p = 0,022).
Byl shromážděn počet subjektů, které podstoupily kolektomii kdykoli během 54 týdnů po první infuzi studijního prostředku, a zkombinovány s údaji ze studií ACT 1 a ACT 2 a jejich rozšíření. Méně subjektů podstoupilo kolektomii v infliximabu 5 mg /kg skupiny (28/242 nebo 11,6% [NS]) a ve skupině infliximabu 10 mg/kg (18/242 nebo 7,4% [p = 0,011]) ve srovnání se skupinou s placebem (36/244; 14,8%).
Snížení výskytu kolektomií bylo také zkoumáno v jiné randomizované, dvojitě zaslepené studii (C0168Y06) u hospitalizovaných pacientů (n = 45) se středně těžkou až těžkou aktivní ulcerózní kolitidou, kteří nereagovali na intravenózní kortikosteroidy. vysoké riziko kolektomie. Do 3 měsíců po infuzi bylo signifikantně méně kolektomií u pacientů, kteří dostali jednu dávku infliximabu v dávce 5 mg / kg, ve srovnání s pacienty, kteří dostávali placebo (29,2 % vs. 66,7 %, p = 0,017).
Ve studiích ACT 1 a ACT 2 zlepšoval infliximab kvalitu života, což bylo potvrzeno statisticky významným zlepšením jak v parametru specifického parametru onemocnění, IBDQ, tak ve zlepšení 36 generických otázek, které tvoří SF-36.
Ankylozující spondylitida u dospělých
Účinnost a bezpečnost infliximabu byla studována ve dvou dvojitě zaslepených, placebem kontrolovaných multicentrických studiích u pacientů s aktivní ankylozující spondylitidou (Bath Index of Ankylosing Spondylitis Disease Activity Activity [BASDAI] score ≥ 4 and pain spinal ≥ 4 on a scale of 1 to 10).
V první studii (P01522), která zahrnovala 3měsíční dvojitě zaslepenou fázi, bylo 70 pacientů léčeno buď infliximabem 5 mg / kg nebo placebem v týdnech 0, 2, 6 (35 pacientů na skupinu). Od 12. týdne začali pacienti dosud léčení placebem dostávat infliximab v dávce 5 mg / kg každých 6 týdnů až do 54. týdne. Po prvním roce bylo 53 pacientů zařazeno do otevřeného protokolu do 102. týdne.
Ve druhé klinické studii (ASSERT) bylo 279 pacientů randomizováno k léčbě placebem (skupina 1, n = 78) nebo infliximabem 5 mg / kg (skupina 2, n = 201) v týdnech 0, 2, 6 a každých 6 týdnů až do 24. týdne. Poté všichni účastníci studie pokračovali v podávání infliximabu každých 6 týdnů až do 96. týdne. Skupina 1 dostala dávku infliximabu 5 mg / kg. Ve skupině 2, počínaje 36. týdnem, byli pacienti, kteří měli BASDAI ≥ 3 po 2 po sobě jdoucí návštěvy, léčeni dávkou infliximabu 7,5 mg / kg každých 6 týdnů až 96. týden.
Ve studii ASSERT bylo zlepšení příznaků a symptomů pozorováno od 2. týdne. Ve 24. týdnu byl počet pacientů, kteří měli odpověď ASAS 20, 15/78 (19%) ve skupině s placebem a roven 123/201 (61 %) ve skupině s infliximabem 5 mg / kg (str
Ve studii P01522 bylo zlepšení známek a symptomů pozorováno od 2. týdne. Ve 12. týdnu byli pacienti, kteří měli odpověď BASDAI 50, 3/35 (9 %) ve skupině s placebem a 20/35 (57 %) v infliximabu 5 skupina mg / kg (str
V obou studiích se výrazně zlepšila fyzická funkce a kvalita života měřená pomocí BASFI a skóre fyzické složky na stupnici SF-36.
Psoriatická artritida u dospělých
Účinnost a bezpečnost byla hodnocena ve dvou dvojitě zaslepených, placebem kontrolovaných, multicentrických studiích u pacientů s aktivní psoriatickou artritidou.
V první klinické studii (IMPACT) byla účinnost a bezpečnost infliximabu studována na 104 pacientech s polyartikulární aktivní psoriatickou artritidou. Během 16týdenní dvojitě zaslepené fáze dostávali pacienti buď 5 mg / kg infliximabu, nebo palcebo. 0, 2, 6 a 14 (52 pacientů v každé skupině). Počínaje 16. týdnem byli pacienti ve skupině s placebem převedeni na infliximab a poté všichni pacienti dostávali infliximab v dávce 5 mg / kg každý. 8 týdnů až týden 46. Po prvním roce studie pokračovalo 78 pacientů s otevřeným prodloužením do 98. týdne.
Ve druhé klinické studii (IMPACT 2) byla účinnost a bezpečnost infliximabu studována na 200 pacientech s aktivní psoriatickou artritidou (oteklé klouby ≥ 5 a bolestivé klouby ≥ 5). 46 % pacientů pokračovalo v fixních dávkách methotrexátu ( ≤ 25 mg / týden) Během 24týdenní dvojitě zaslepené fáze dostávali pacienti buď 5 mg / kg infliximabu, nebo placebo v týdnech 0, 2, 6, 14 a 22 (100 pacientů v každé skupině). V 16. týdnu 47 pacientů dostávalo placebo se zlepšením
Klíčové výsledky účinnosti pro IMPACT a IMPACT 2 jsou uvedeny níže
Tabulka 9
Účinky na ACR a PASI v IMPACT a IMPACT 2
* Analýza ITT, do které byly zahrnuty subjekty s chybějícími údaji jako Ne-respondenti
v týdnu 98 údaje IMPACT zahrnují pacienty ze skupiny s placebem a pacienty léčené infliximabem, kteří vstoupili do otevřeného prodloužení
b Na základě pacientů s PASI ≥ 2,5 na počátku pro IMPACT a pacientů s postižením psoriatického povrchu těla (BSA) na počátku ≥ 3% v IMPACT 2
** Odpověď PASI 75 pro IMPACT není zahrnuta kvůli nízkému N; p
U IMPACT a IMPACT 2 byla klinická odpověď pozorována již ve 2. týdnu a byla udržována do 98. týdne, respektive 54. týdne. Účinnost byla prokázána se souběžným užíváním methotrexátu i bez něj. U pacientů léčených infliximabem bylo pozorováno snížení parametrů periferní aktivity charakteristické pro psoriatickou artritidu (jako je počet oteklých kloubů, počet bolestivých / citlivých kloubů, daktylitida a přítomnost entezopatií).
Radiografické změny byly hodnoceny v IMPACT2. Rentgenové snímky rukou a nohou byly shromážděny na začátku, 24. a 54. týden. Léčba infliximabem snížila rychlost progrese poškození periferních kloubů ve srovnání s léčbou placebem v primárním koncovém bodě 24. týdne, měřeno jako změna od výchozího stavu v modifikovaném celkovém skóre vdH -S (průměr ± SD skóre bylo 0,82 ± 2,62 ve skupině s placebem oproti - 0,70 ± 2,53 ve skupině s infliximabem; p
Významné zlepšení tělesných funkcí hodnocené pomocí HAQ bylo prokázáno u pacientů léčených infliximabem a bylo také prokázáno významné zlepšení kvality života měřené souhrnným skóre fyzických a mentálních složek SF-36 v IMPACT 2.
Psoriáza u dospělých
Účinnost infliximabu byla hodnocena ve dvou randomizovaných, dvojitě zaslepených, multicentrických studiích, SPIRIT a EXPRESS. Pacienti v obou studiích měli ložiskovou psoriázu (BSA [Body Surface Area] ≥ 10% a PASI skóre [Psoriasis Area and Severity Index] ≥ 12 ) Primárním cílovým parametrem v obou studiích byl podíl pacientů, kteří dosáhli ≥ 75% zlepšení oproti výchozímu skóre PASI v 10. týdnu.
SPIRIT vyhodnotil účinnost indukční terapie infliximabem u 249 pacientů s ložiskovou psoriázou dříve léčených PUVA nebo systémovou terapií.Pacienti dostávali infuze 3 nebo 5 mg / kg infliximabu nebo placeba v týdnech 0, 2 a 6. Pacienti s PGA ≥ 3 měli nárok na další infuzi stejné léčby ve 26. týdnu.
Ve skupině SPIRIT byl podíl pacientů, kteří dosáhli PASI 75 v 10. týdnu, 71,7% ve skupině s infliximabem 3 mg / kg, 87,9% ve skupině s infliximabem 5 mg / kg a ve skupině s infliximabem 5 mg / kg. 5,9% ve skupině skupina s placebem (p 20 týdnů. Nebyly pozorovány žádné rebound fenomény.
Společnost EXPRESS hodnotila účinnost indukční a udržovací terapie infliximabem u 378 pacientů s ložiskovou psoriázou. Pacienti dostávali infuze 5 mg / kg infliximabu nebo placebo v týdnech 0, 2 a 6, po nichž následovalo každých 8 udržovacích terapií. Týdny až 22. týden ve skupině s placebem a do 46. týdne ve skupině s infliximabem. V 24. týdnu skupina s placebem přešla na indukční terapii infliximabem (5 mg / kg), po níž následovala udržovací terapie infliximabem (5 mg / kg) Psoriáza na nehtech byla hodnocena pomocí indexu závažnosti psoriázy na nehtech (NAPSI) ) Předchozí terapii PUVA, methotrexátem, cyklosporinem nebo acitretinem obdrželo 71,4% pacientů, ačkoli tito nebyli nutně rezistentní na terapii. Nejvýznamnější výsledky jsou uvedeny v tabulce 10. U subjektů léčených infliximabem byly významné odpovědi na PASI 50 evidentní v prvním v isita (2. týden) a reakce na PASI 75 při druhé návštěvě (6. týden). Účinnost v podskupině pacientů, kteří dříve podstoupili systémovou terapii, byla podobná jako v celkové populaci studie.
Tabulka 10
Souhrn odpovědí PASI, odpovědí PGA a procenta pacientů se všemi nehty uzdravenými v 10., 24. a 50. týdnu. EXPRESS.
na str
b n = 292
c Analýza byla provedena u subjektů s psoriázou nehtů na počátku (81,8% subjektů). Průměrné výchozí skóre NAPSI bylo 4,6 a 4,3 ve skupině s infliximabem a placebem.
Významná vylepšení oproti výchozím hodnotám byla patrná u DLQI (str
Pediatrická populace
Crohnova choroba u pediatrických pacientů (6-17 let)
Ve studii REACH bylo 112 pacientů (ve věku 6-17 let, průměrný věk 13 let) se středně těžkou až těžkou aktivní Crohnovou chorobou (pediatrický průměr CDAI 40) a s nedostatečnou odpovědí na konvenční terapie, léčeno 5 mg / kg infliximab v týdnech 0, 2 a 6. U všech pacientů byla vyžadována stabilní dávka 6-MP, AZA nebo MTX (35% bylo na začátku také na kortikosteroidech). Pacienti, u nichž vyšetřovatel považoval klinickou odpověď v 10. týdnu, byli poté randomizováni do dvou skupin a dostávali infliximab 5 mg / kg každých 8 týdnů nebo každých 12 týdnů jako udržovací léčbu. Pokud došlo během udržovací léčby ke ztrátě odpovědi, bylo možné přejít na vyšší dávku (10 mg / kg) a / nebo v kratších intervalech mezi infuzemi (každých 8 týdnů). Tento přechod podstoupilo 32 dětských pacientů hodnotitelných pro účely studie (9 subjektů ve skupině léčených každých 8 týdnů a 23 subjektů ve skupině léčených každých 12 týdnů). Dvacet čtyři z těchto pacientů (75,0%) získalo klinickou odpověď po tomto přechodu.
Procento pacientů s klinickou odpovědí v týdnu 10 bylo 88,4% (99/112). Procento subjektů dosahujících klinické remise v týdnu 10 bylo 58,9% (66/112).
Ve 30. týdnu bylo procento pacientů v klinické remisi vyšší ve skupině každých 8 týdnů (59,6%, 31/52) než u pacientů v udržovací skupině každých 12 týdnů (35,3%, 18/51; p = 0,013) . V 54. týdnu byla data následující: 55,8% (29/52) v udržovací skupině léčené každých 8 týdnů a 23,5% (12/51) ve udržovací skupině léčené každých 12 týdnů (p
Data o píštělích byla extrahována ze skóre PCDAI. Z 22 pacientů, kteří měli na počátku píštěle, 63,6% (14/22), 59,1% (13/22) a 68,2% (15/22) odpovědělo na píštěl úplně. V 10., 30. a 54, respektive s ohledem na celkově udržovací skupiny, jak ty, které byly léčeny každých 8 týdnů, tak ty, které byly léčeny každých 12 týdnů.
Kromě toho bylo od počátku pozorováno statisticky a klinicky významné zlepšení kvality života a výšky a také významné snížení používání kortikosteroidů.
Dětská ulcerózní kolitida (6-17 let)
Bezpečnost a účinnost infliximabu byla hodnocena v multicentrické, randomizované, otevřené, paralelní skupině klinických studií (C0168T72) u 60 pediatrických pacientů ve věku 6-17 let (medián věku 14,5 let) s kolitidou. Středně těžká až těžká aktivní vředová choroba ( Mayo skóre 6 až 12; endoskopické podskóre ≥ 2) s neadekvátní odpovědí na konvenční terapie.Na začátku 53% pacientů dostávalo imunomodulační terapii (6-MP, AZA a / nebo MTX) a 62% pacientů dostávalo kortikosteroidy. po týdnu 0 byly povoleny imunomodulátory a snižování dávky kortikosteroidů.
Všichni pacienti dostali indukční režim infliximabu 5 mg / kg v týdnech 0, 2 a 6. Pacienti, kteří v 8. týdnu (n = 15) nereagovali na infliximab, nedostali žádné léky a vrátili se k následnému hodnocení bezpečnosti. V 8. týdnu bylo randomizováno 45 pacientů, kteří dostávali udržovací léčbu infliximabem 5 mg / kg každých 8 týdnů nebo každých 12 týdnů.
Podíl pacientů s klinickou odpovědí v 8. týdnu byl 73,3% (44/60). Klinická odpověď v 8. týdnu byla u pacientů s výchozím stavem a bez současného použití imunomodulátorů podobná. Klinická remise v 8. týdnu byla 33,3% (17/51), měřeno podle skóre indexu aktivity pediatrické ulcerózní kolitidy (PUCAI).
V 54. týdnu byl podíl pacientů v klinické remisi měřený skóre PUCAI 38% (8/21) ve skupině s 8týdenní udržovací léčbou a 18% (4/22) v každé 12týdenní udržovací skupině. . U pacientů užívajících na začátku léčby kortikosteroidy byl podíl pacientů v remisi a nedostávajících kortikosteroidy v 54. týdnu 38,5% (5/13) ve skupině s udržovací léčbou každých 8 týdnů a 0% (0/13) ve skupině s udržovací léčbou každých 12 týdny.
V této studii bylo více pacientů ve skupině 12 až 17 let než ve skupině 6 až 11 let (45/60 vs. 15/60). Ačkoli počet pacientů v každé podskupině byl příliš malý na to, aby bylo možné vyvodit pevné závěry týkající se „vlivu věku“, v mladší věkové skupině byl vyšší počet pacientů, kteří kvůli nedostatečné účinnosti zvýšili dávku nebo přerušili léčbu.
Jiné pediatrické indikace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Remicade u všech podskupin pediatrické populace u revmatoidní artritidy, juvenilní idiopatické artritidy, psoriatické artritidy, ankylozující spondylitidy, psoriázy a Crohnovy choroby (informace o použití u dětí viz bod 4.2) ).
05,2 "Farmakokinetické vlastnosti
Jednorázové intravenózní infuze 1, 3, 5, 10 nebo 20 mg / kg infliximabu zvýšily jak maximální sérové koncentrace (Cmax), tak plochu pod křivkou závislosti koncentrace na čase (AUC) v závislosti na dávce. Distribuce v ustáleném stavu (medián Vd 3,0-4,1 litru) byl nezávislý na podané dávce, což ukazuje, že infliximab je distribuován hlavně do vaskulárního kompartmentu. Nebyla pozorována žádná časová závislost farmakokinetických charakteristik. infliximab nebyl charakterizován. Nemodifikovaný infliximab nebyl nalezen v moči. Ne u pacientů s revmatoidní artritidou byly pozorovány velké rozdíly v clearance nebo distribučním objemu související s věkem nebo hmotností. Farmakokinetika infliximabu u starších pacientů nebyla studována. U pacientů s poruchou funkce jater nebo ledvin nebyly provedeny žádné studie.
Při jednorázových dávkách 3, 5 nebo 10 mg / kg byly průměrné hodnoty Cmax 77, 118 a 277 mcg / ml. Průměrný terminální poločas při těchto dávkách se pohyboval od 8 do 9,5 dne. U většiny pacientů při doporučené jednorázové dávce 5 mg / kg u Crohnovy choroby a 3 mg / kg každých 8 týdnů k udržení. U revmatoidní artritidy mohl infliximab být detekovány v séru po dobu nejméně 8 týdnů.
Opakované podávání infliximabu (5 mg / kg v týdnech 0, 2 a 6 při fistulizující Crohnově chorobě, 3 nebo 10 mg / kg každé 4 nebo 8 týdnů při revmatoidní artritidě) mělo za následek mírné nahromadění infliximabu v séru po druhé dávce Žádné další byla pozorována klinicky relevantní akumulace U většiny pacientů s fistulizující Crohnovou chorobou byl infliximab detekován v séru po dobu 12 týdnů (rozmezí 4–28 týdnů) po podání režimu.
Pediatrická populace
„Populační farmakokinetická analýza založená na údajích získaných od pacientů s ulcerózní kolitidou (N = 60), Crohnovou chorobou (N = 112), juvenilní revmatoidní artritidou (N = 117) a Kawasakiho chorobou (N = 16) ve věku od 2 měsíců do 17 let celkově ukázalo, že expozice infliximabu byla nelineárně závislá na tělesné hmotnosti. Po podání 5 mg / kg přípravku Remicade každých 8 týdnů byla očekávaná střední expozice infliximabu v ustáleném stavu (plocha pod křivkou koncentrace-čas v ustáleném stavu, AUCss) u pediatrických pacientů ve věku 6 až 17 let přibližně o 20% nižší než medián předpokládané expozice léčiva v ustáleném stavu u dospělých. Medián AUCss u pediatrických pacientů ve věku 2 až méně než 6 let byl předpovídán přibližně o 40% nižší než u dospělých, i když počet pacientů podporujících tento odhad je omezený.
05.3 Předklinické údaje vztahující se k bezpečnosti
Infliximab nereaguje zkříženě s TNFα u jiných živočišných druhů než lidí a šimpanzů. Proto jsou konvenční preklinické údaje o bezpečnosti infliximabu omezené. Ve studii vývojové toxicity u myší s použitím podobné protilátky, která selektivně inhibuje funkční aktivitu myšího TNFα, nebyla zjištěna mateřská toxicita, embryotoxicita, teratogenita. Ve studii fertility a obecné reprodukční funkce byl počet březích myší po podání stejné analogické protilátky snížen. Není známo, zda tyto nálezy byly způsobeny účinky na muže a / nebo ženy.V 6měsíční studii toxicity po opakovaných dávkách u potkanů, za použití stejných analogických protilátek proti myšímu TNFa, byly pozorovány krystalické usazeniny na pouzdru čočky některých ošetřených samců potkanů. U pacientů nebylo provedeno žádné specifické oftalmologické vyšetření k posouzení relevance této události u lidí. K hodnocení karcinogenního potenciálu infliximabu nebyly provedeny dlouhodobé studie. Ve studiích prováděných na myších s deficitem TNFa bylo ukázáno, že nedochází k nárůstu nádorů, pokud je vyvolán známými iniciátory nádorů a / nebo promotory.
06.0 FARMACEUTICKÉ INFORMACE
06.1 Pomocné látky
Sacharóza
Polysorbát 80
Monobazický fosforečnan sodný
Difázický fosforečnan sodný
06.2 Neslučitelnost
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
06.3 Doba platnosti
Před rekonstitucí:
3 roky při 2 ° C - 8 ° C
Remicade může být uchováván při teplotách nepřesahujících 25 ° C po dobu jednoho období až 6 měsíců, ale ne po původním datu expirace. Na krabici musí být napsáno nové datum vypršení platnosti. Po vyjmutí z chladničky by neměl být přípravek Remicade znovu uchováván v chladničce.
Po rekonstituci:
Chemická a fyzikální stabilita naředěného roztoku byla prokázána po dobu 24 hodin při 25 ° C. Z mikrobiologického hlediska by měl být přípravek použit co nejdříve a v každém případě do 3 hodin po rozpuštění a naředění. . Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a v žádném případě by neměl překročit 24 hodin při 2 ° C až 8 ° C.
06.4 Zvláštní opatření pro skladování
Uchovávejte v chladničce (2 ° C - 8 ° C).
Podmínky uchovávání do 25 ° C před rekonstitucí léčivého přípravku viz bod 6.3.
Podmínky uchovávání po rekonstituci léčivého přípravku viz bod 6.3.
06.5 Charakter vnitřního obalu a obsah balení
Injekční lahvička ze skla typu I s gumovou zátkou a hliníkovým lemováním chráněná plastovým víčkem, obsahující 100 mg infliximabu.
Remicade je k dispozici v balení po 1, 2, 3, 4 nebo 5 injekčních lahvičkách. Na trhu nemusí být všechny velikosti balení.
06.6 Návod k použití a zacházení
1. Vypočítejte dávku a počet potřebných injekčních lahviček Remicade. Jedna injekční lahvička přípravku Remicade obsahuje 100 mg infliximabu. Vypočítejte celkový požadovaný objem rekonstituovaného roztoku Remicade.
2. Za aseptických podmínek rekonstituujte každou injekční lahvičku Remicade 10 ml vody na injekci pomocí injekční stříkačky s jehlou velikosti 21 (0,8 mm) nebo menší. Sejměte hliníkový jazýček z lahvičky a očistěte víčko vatovým tamponem namočeným v 70% alkoholu. Vložte jehlu injekční stříkačky do injekční lahvičky středem gumové zátky a nasměrujte tok vody na injekci na skleněnou stěnu injekční lahvičky. Roztokem jemně zatočte, aby se lyofilizovaný prášek úplně rozpustil. Netřepejte prudce nebo dlouhodobě Během rekonstituce může dojít k pěnění. Rekonstituovaný roztok nechte 5 minut odstát. Zkontrolujte, zda je roztok bezbarvý až žlutý a opalescentní, roztok může obsahovat malé průsvitné částice, protože infliximab je protein. roztok, pokud si všimnete matných částic, změny barvy nebo jiných cizích těles.
3. Zřeďte celkový objem dávky rekonstituovaného roztoku Remicade na 250 ml pomocí infuzního roztoku chloridu sodného 9 mg / ml (0,9%). Rekonstituovaný roztok Remicade neřeďte žádným jiným ředidlem. To lze provést odebráním objemu infuzního roztoku chloridu sodného 9 mg / ml (0,9%) ze 250 ml skleněné lahve nebo infuzního vaku, který se rovná objemu rekonstituovaného přípravku Remicade. Pomalu přidejte celkový objem rekonstituovaného roztoku Remicade do 250 ml lahve nebo infuzního vaku. Jemně promíchejte.
4. Infuzní roztok podávejte po dobu infuze, která není kratší než doporučená doba infuze (viz bod 4.2). Používejte pouze infuzní soupravu se sterilním, nepyrogenním in-line filtrem s nízkou vazbou bílkovin (průměr póru 1,2 mikrometru nebo méně). Jelikož neobsahuje žádné konzervační látky, doporučuje se zahájit podávání roztoku pro intravenózní infuzi co nejdříve a do 3 hodin od rekonstituce a naředění. Pokud se rekonstituce a ředění provádí za aseptických podmínek, lze infuzní roztok Remicade použít do 24 hodin, pokud je uchováván při teplotě 2 ° C až 8 ° C. Nepoužitý roztok by neměl být uchováván pro pozdější použití.
5. Studie fyzikální a biochemické kompatibility nebyly provedeny za účelem vyhodnocení kombinace přípravku Remicade s jinými činidly. Nepodávejte Remicade současně s jinými léčivými přípravky ve stejné intravenózní linii.
6. Před podáním vizuálně zkontrolujte Remicade, abyste se ujistili, že nejsou pozorovány žádné částice nebo změna barvy.Pokud jsou pozorovány neprůhledné částice, změna barvy nebo cizí částice, nepoužívejte.
7. Nepoužitý lék a odpady z tohoto léčivého přípravku musí být zlikvidovány v souladu s místními předpisy.
07.0 DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen Biologics B.V. Einsteinweg 101
2333 CB Leiden
Holandsko
08.0 REGISTRAČNÍ ČÍSLO
EU/1/99/116/001
034528012
EU/1/99/116/002
EU/1/99/116/003
EU/1/99/116/004
EU/1/99/116/005
09.0 DATUM PRVNÍ REGISTRACE NEBO PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. srpna 1999. Datum posledního prodloužení: 2. července 2009.
10.0 DATUM REVIZE TEXTU
24. září 2015












.jpg)






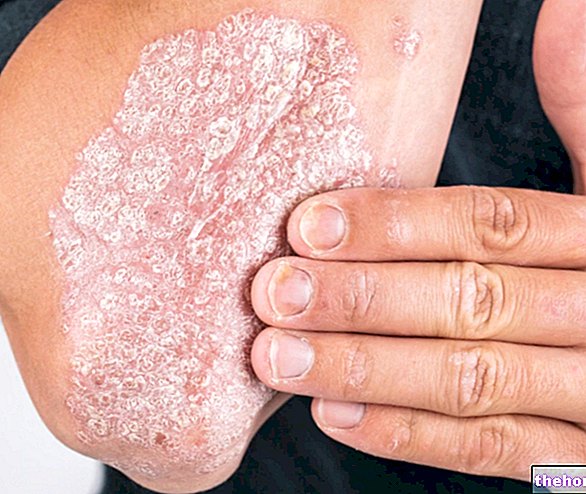
.jpg)

-cos-cause-e-disturbi-associati.jpg)
