Všeobecnost
Mitochondriální DNA nebo mtDNA je deoxyribonukleová kyselina, která sídlí uvnitř mitochondrií, tj. Organel eukaryotických buněk odpovědných za velmi důležitý buněčný proces oxidativní fosforylace.

Má však také některé zvláštnosti, strukturální i funkční, díky nimž je jedinečný svého druhu. Mezi tyto zvláštnosti patří: kruhovitost dvojvlákna nukleotidů, obsah genů (což je pouze 37 prvků) a téměř úplná absence nekódujících nukleotidových sekvencí.
Mitochondriální DNA plní základní funkci pro přežití buněk: produkuje enzymy nezbytné pro realizaci oxidativní fosforylace.
Co je mitochondriální DNA?
Mitochondriální DNA nebo mtDNA je DNA umístěná v mitochondriích.
Mitochondrie jsou velké buněčné organely, typické pro eukaryotické organismy, které přeměňují chemickou energii obsaženou v potravě na ATP, což je forma energie, kterou mohou buňky využívat.
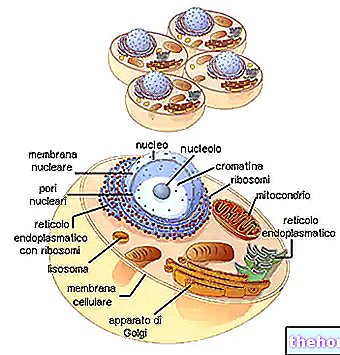
POZADÍ STRUKTURY A FUNKCÍ MITOCHONDRONŮ
Mitochondrie mají tubulární, vláknitý nebo zrnitý tvar a nacházejí se v cytoplazmě a zabírají téměř 25% jejího objemu.
Mají dvě fosfolipidové dvojvrstvy, jednu vnější a druhou vnitřní.
Nejvzdálenější membrána, známá jako vnější mitochondriální membrána, představuje obvod každého mitochondrií a má transportní proteiny (poriny a další), díky nimž je propustná pro molekuly o velikosti rovné nebo menší než 5 000 daltonů.
Nejvnitřnější membrána, známá jako vnitřní mitochondriální membrána, obsahuje všechny enzymatické (nebo enzymatické) a koenzymové složky, nezbytné pro syntézu ATP, a definuje centrální prostor, nazývaný matice.
Na rozdíl od nejvzdálenější membrány má vnitřní mitochondriální membrána četné invaginace - takzvané hřebeny - které zvětšují její celkovou plochu.
Mezi dvěma mitochondriálními membránami je prostor téměř 60-80 angströmů (A). Tento prostor se nazývá intermembránový prostor. Mezimembránový prostor má složení velmi podobné složení cytoplazmy.
Syntéza ATP, provozovaná mitochondriemi, je velmi složitý proces, který biologové ztotožňují s pojmem oxidativní fosforylace.
PŘESNÉ UMÍSTĚNÍ MITOCHONDRÁLNÍ DNA A MNOŽSTVÍ

Obrázek: lidský mitochondrion.
Mitochondriální DNA sídlí v mitochondriální matrici, tj. V prostoru vymezeném vnitřní mitochondriální membránou.
Podle spolehlivých vědeckých studií může každý mitochondrion obsahovat 2 až 12 kopií mitochondriální DNA.
Vzhledem k tomu, že v lidském těle mohou některé buňky obsahovat několik tisíc mitochondrií, může celkový počet kopií mitochondriální DNA v jedné lidské buňce dosáhnout až 20 000 jednotek.
Vezměte prosím na vědomí: počet mitochondrií v lidských buňkách se liší v závislosti na typu buňky. Například hepatocyty (tj. Jaterní buňky) mohou obsahovat mezi 1 000 a 2 000 mitochondrií, zatímco erytrocyty (tj. Červené krvinky) je zcela postrádají.
Struktura
Obecná struktura mitochondriální molekuly DNA se podobá obecné struktuře jaderné DNA, tj. Genetickému dědictví přítomnému v jádru eukaryotických buněk.
Vskutku, analogicky k jaderné DNA:
- Mitochondriální DNA je biopolymer, skládající se ze dvou dlouhých řetězců nukleotidů. Nukleotidy jsou organické molekuly, které jsou výsledkem spojení tří prvků: cukru s 5 atomy uhlíku (v případě DNA deoxyribózy), dusíkaté báze a fosfátové skupiny.
- Každý nukleotid mitochondriální DNA se váže na další nukleotid stejného řetězce pomocí fosfodiesterové vazby mezi uhlíkem 3 jeho deoxyribózy a fosfátovou skupinou bezprostředně následujícího nukleotidu.
- Dvě vlákna mitochondriální DNA mají opačnou orientaci, přičemž konec jednoho interaguje s koncem druhého a naopak. Toto konkrétní uspořádání je známé jako antiparalelní uspořádání (nebo antiparalelní orientace).
- Dvě vlákna mitochondriální DNA na sebe vzájemně působí prostřednictvím dusíkatých bází.
Konkrétně každá dusíkatá báze každého vlákna vytváří vodíkové vazby s jednou a pouze jednou dusíkatou zásadou přítomnou na druhém vláknu.
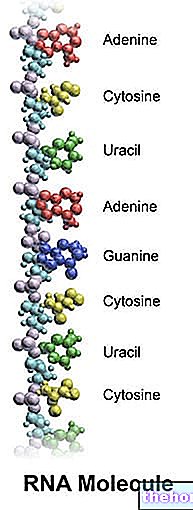
Tento typ interakce se nazývá „párování mezi dusíkatými bázemi“ nebo „pár dusíkatých bází“. - Dusíkaté báze mitochondriální DNA jsou adenin, tymin, cytosin a guanin.
Párování, ke kterému tyto dusíkaté báze vedou, není náhodné, ale vysoce specifické: adenin interaguje pouze s thyminem, zatímco cytosin interaguje pouze s guaninem. - Mitochondriální DNA je domovem genů (nebo genových sekvencí). Geny jsou sekvence více či méně dlouhých nukleotidů s dobře definovaným biologickým významem. Ve většině případů dávají vzniknout bílkovinám.

STRUKTURÁLNÍ ÚDAJE MITOCHONDRÁLNÍ DNA
Kromě výše uvedených analogií má lidská mitochondriální DNA některé strukturální zvláštnosti, které ji výrazně odlišují od lidské jaderné DNA.
Za prvé je to kruhová molekula, zatímco jaderná DNA je lineární molekula.
Má tedy 16 569 párů dusíkatých bází, zatímco jaderná DNA má ohromných 3,3 miliardy.
Obsahuje 37 genů, zatímco jaderná DNA podle všeho obsahuje 20 000 až 25 000.
Není organizována v chromozomech, zatímco jaderná DNA je rozdělena na 23 chromozomů a forem, s některými specifickými proteiny, látkou zvanou chromatin.
Nakonec obsahuje řadu nukleotidů, které se účastní současně dvou genů, zatímco jaderná DNA má geny, jejichž nukleotidové sekvence jsou dobře definované a navzájem odlišné.
Původ
Mitochondriální DNA má s největší pravděpodobností „bakteriální“ původ.
Na základě četných nezávislých studií se molekulární biologové domnívají, že buněčná přítomnost mitochondriální DNA je výsledkem začlenění nezávislých bakteriálních organismů rodovými eukaryotickými buňkami, velmi podobných mitochondriím.
Tento kuriózní objev ohromil vědeckou komunitu jen částečně, protože DNA přítomná v bakteriích je obecně kruhový nukleotidový řetězec, jako mitochondriální DNA.
Teorie, podle níž mají mitochondrie a mitochondriální DNA „bakteriální původ, přebírá název„ endosymbiotická teorie “od slova„ endosymbióza “. Stručně, v biologii termín„ endosymbióza “označuje spolupráci mezi dvěma organismy, která zahrnuje „začlenění jednoho do druhého, za účelem získání určité výhody.
Zvědavost
Podle spolehlivých vědeckých studií by v průběhu evoluce mnoho bakteriálních genů, přítomných na budoucí mitochondriální DNA, změnilo umístění a přesunulo by se do jaderné DNA.
Jinými slovy, na začátku endosymbiózy některé geny nyní přítomné v jaderné DNA sídlily v DNA těchto bakteriálních organismů, z nichž se později staly mitochondrie.
Na podporu teorie týkající se posunu genů mezi mitochondriální DNA a jadernou DNA je pozorování, že některé geny pocházejí z mitochondriální DNA u některých druhů a od jaderné DNA u jiných.
Funkce
Mitochondriální DNA produkuje enzymy (tj. Proteiny), nezbytné pro správnou implementaci delikátního procesu oxidativní fosforylace.
Pokyny pro syntézu těchto enzymů se nacházejí v 37 genech, které tvoří genom mitochondriální DNA.
JAKÝ MITOCHONDRÁLNÍ DNA GENY KÓD: PODROBNOSTI
37 genů mitochondriální DNA kóduje: proteiny, tRNA a rRNA.
Zejména:
- 13 kóduje 13 proteinů odpovědných za provádění oxidační fosforylace
- 22 kód pro 22 molekul tRNA
- 2 kódují 2 molekuly rRNA
Molekuly tRNA a rRNA jsou zásadní pro syntézu výše uvedených 13 proteinů, protože tvoří strojní zařízení, které reguluje jejich produkci.
Jinými slovy, mitochondriální DNA má informace k produkci určité sady proteinů a nástrojů nezbytných pro jejich syntézu.
Co jsou RNA, tRNA a rRNA?
RNA nebo ribonukleová kyselina je nukleová kyselina, která hraje zásadní roli při generování proteinů, počínaje DNA.
Obecně ANN může existovat v různých formách (nebo typech) v závislosti na konkrétní funkci, na kterou je delegována.
TRNA a rRNA jsou dvě z těchto možných forem.
TRNA se používá k přidávání aminokyselin během procesu výroby proteinů.Aminokyseliny jsou molekulární jednotky, které tvoří bílkoviny.
RRNA tvoří ribozomy, tj. Buněčné struktury, ve kterých probíhá syntéza proteinů.
Chcete -li podrobně poznat ANN a jeho funkce, mohou čtenáři kliknout sem.
FUNKČNÍ PODROBNOSTI O MITOCHONDRÁLNÍ DNA
Z funkčního hlediska má mitochondriální DNA některé zvláštní vlastnosti, které ji jasně odlišují od jaderné DNA.
Z čeho se tyto zvláštní vlastnosti skládají:
- Mitochondriální DNA je polonezávislá v tom smyslu, že potřebuje zásah některých proteinů syntetizovaných z jaderné DNA.
Na druhé straně je jaderná DNA zcela autonomní a sama produkuje vše, co potřebuje ke správnému plnění svých úkolů. - Mitochondriální DNA má mírně odlišný genetický kód než jaderná DNA. To vede k řadě rozdílů ve výrobě proteinů: pokud určitá sekvence nukleotidů v jaderné DNA vede k vytvoření určitého proteinu, vede stejná sekvence v mitochondriální DNA ke vzniku mírně odlišného proteinu.
- Mitochondriální DNA má velmi málo nekódujících nukleotidových sekvencí, to znamená, že neprodukují žádné proteiny, tRNA ani rRNA. V procentech jsou nekódující pouze 3% mitochondriální DNA.
Na druhou stranu jaderná DNA kóduje pouze 7%, takže obsahuje mnoho nekódujících nukleotidových sekvencí (celých 93%).
Tabulka: shrnutí rozdílů mezi lidskou mitochondriální DNA a lidskou jadernou DNA.
Mitochondriální DNA
Jaderná DNA
- Je kruhový
- Je lineární
- Má celkem 16 569 párů dusíkatých bází
- Má celkem 3,3 miliardy párů dusíkatých bází
- Obsahuje celkem 37 genů
- Obsahuje 20 000 až 25 000 genů
- Ke správnému fungování potřebuje podporu některých genových produktů, pocházejících z jaderné DNA
- Je autonomní a sám produkuje vše, co potřebuje ke správnému výkonu svých funkcí
- Může být přítomen v několika kopiích v rámci každé jednotlivé mitochondrie
- Je jedinečný, to znamená, že je pouze v jedné kopii a sídlí v jádru
- Kóduje 97% nukleotidové sekvence, která ji tvoří
- Pouze 7% nukleotidové sekvence, která ji tvoří, kóduje
- Není organizován do chromozomů
- Je rozdělena na 23 chromozomů
- Používá genetický kód mírně odlišný od toho, takříkajíc „tradičního“
- Použijte „tradiční“ genetický kód
- Jeho dědičnost je mateřská
- Jeho dědičnost je napůl mateřská a napůl otcovská
- Některé z jeho nukleotidů se účastní dvou genů současně
- Sekvence nukleotidů, které tvoří geny, jsou navzájem dobře odlišeny
Dědictví
Mitochondriální dědičnost DNA je čistě mateřská.
To znamená, že v páru rodičů je to žena, která přenáší mitochondriální DNA na potomstvo (tj. Na děti).
Zcela opačným způsobem, než je uvedeno výše, je dědičnost jaderné DNA napůl mateřská a napůl otcovská. Jinými slovy, oba rodiče přispívají rovnoměrně k přenosu jaderné DNA u potomků.
Vezměte prosím na vědomí: mateřská dědičnost mitochondriální DNA zahrnuje také mitochondriální strukturu. Mitochondrie přítomné u jednotlivce jsou tedy mateřské.
Související patologie
Předpoklad: Genetická mutace je trvalá změna v sekvenci nukleotidů, které tvoří jaderný nebo mitochondriální gen DNA.
Typicky má přítomnost genetické mutace za následek „změnu nebo ztrátu normální funkce příslušného genu.
Přítomnost mutací v genech mitochondriální DNA může vést k celé řadě onemocnění, včetně:
- Leberova dědičná optická neuropatie
- Kearns-Sayreův syndrom
- Leighův syndrom
- Nedostatek cytochrom C oxidázy
- Progresivní vnější oftalmoplegie
- Pearsonův syndrom
- Mitochondriální encefalomyopatie s laktátovou acidózou a epizodami podobnými mrtvici (syndrom MELAS)
- Diabetes s mateřsky přenosnou hluchotou
- Myoklonická epilepsie s nepravidelnými červenými vlákny
Pokud jde o patologické stavy spojené s jednou nebo více mitochondriálními mutacemi DNA, je třeba vyjasnit dva aspekty.
Za prvé, závažnost onemocnění závisí na kvantitativním vztahu mezi mutovanými mitochondriálními DNA a zdravými, normální mitochondriální DNA. Pokud je počet mutovaných mitochondriálních DNA výrazně vyšší než u zdravých DNA, bude výsledný stav vážnější.
Za druhé, mutace v mitochondriální DNA postihují pouze některé tkáně organismu, zejména ty, které vyžadují velké množství ATP vyplývající z procesu oxidační fosforylace. To je celkem pochopitelné: trpět více než jednou poruchou mitochondriální DNA jsou buňky, které to nejvíce potřebují funkce, kterou mitochondriální DNA normálně plní.
LEBEROVA HEREDITÁRNÍ OPTICKÁ NEUROPATIE
Leberova dědičná optická neuropatie vzniká v důsledku mutace až čtyř mitochondriálních genů DNA. Tyto geny obsahují informace, které vedou k syntéze takzvaného komplexu I (nebo NADH oxid-reduktázy), jednoho z různých enzymů zapojených do procesu oxidační fosforylace.
Projevy patologie spočívají v postupné degeneraci zrakového nervu a postupné ztrátě zraku.
KEARNS-SAYRE SYNDROM
Kearns-Sayreův syndrom se objevuje kvůli nedostatku dostatečného množství mitochondriální DNA (poznámka: nedostatek určité nukleotidové sekvence se nazývá delece).
U lidí s Kearns-Sayreovým syndromem se vyvíjí oftalmoplegie (celková nebo částečná paralýza okulomotorických svalů), forma retinopatie a abnormality srdečního rytmu (atrioventrikulární blok).
LEIGHŮV SYNDROM
Leighův syndrom vzniká v důsledku mitochondriálních mutací DNA, které mohou ovlivnit protein ATP-syntázy (také nazývaný V-komplex) a / nebo některé tRNA.
Leighův syndrom je progresivní neurologické onemocnění, které se objevuje v kojeneckém nebo dětském věku a je zodpovědné za: vývojové zpoždění, svalovou slabost, periferní neuropatii, motorické poruchy, dýchací potíže a oftalmoplegii.
DEFICITY CYTOCHROME C OXIDASE
Deficit oxidázy cytochromu C nastává v důsledku mutace alespoň 3 genů mitochondriální DNA. Tyto geny jsou nezbytné pro správnou syntézu enzymu oxidázy cytochromu C (nebo komplexu IV), zapojeného do procesu oxidační fosforylace.
Typické projevy nedostatku oxidázy cytochromu C zahrnují: dysfunkci kosterního svalstva, srdeční dysfunkci, renální dysfunkci a jaterní dysfunkci.
PROGRESIVNÍ VNĚJŠÍ OČNÍ MOPLE
Progresivní vnější oftalmoplegie vzniká z nedostatku podstatného počtu mitochondriálních DNA nukleotidů (delece)
S progresivním charakterem (jak lze usuzovat z názvu) tato patologie způsobuje ochrnutí okulomotorických svalů s následnou ptózou a značnými problémy se zrakem.
PEARSONŮV SYNDROM
Pearsonův syndrom se objevuje po nápadné deleci mitochondriální DNA podobným způsobem jako progresivní vnější oftalmoplegie a Kearns-Sayreův syndrom.
Typické projevy Pearsonova syndromu zahrnují: sideroblastickou anémii, pankreatickou dysfunkci (např. Diabetes závislý na inzulínu), neurologické deficity a svalové poruchy.
Pearsonův syndrom obvykle způsobí smrt postiženého v mladém věku. Ve skutečnosti ti, kterých se tato patologie týká, jen zřídka dosáhnou dospělosti.
MELASOVÝ SYNDROM
Syndrom MELAS, také známý jako mitochondriální encefalomyopatie s laktátovou acidózou a epizodami podobnými mrtvici, vzniká mutací alespoň 5 mitochondriálních DNA genů.
Tyto geny přispívají k syntéze NADH oxid-reduktázy nebo komplexu I a některých tRNA.
Syndrom MELAS zahrnuje přítomnost neurologických poruch, svalových poruch, neobvyklé akumulace kyseliny mléčné v tkáních (se všemi doprovodnými příznaky), problémy s dýcháním, ztrátu kontroly funkce střev, opakující se únavu, problémy s ledvinami, srdeční problémy, cukrovku, epilepsii a nedostatek koordinace.
DALŠÍ PATOLOGIE
Podle různých vědeckých studií by onemocnění, jako je syndrom cyklického zvracení, retinitis pigmentosa, ataxie, Parkinsonova choroba a Alzheimerova choroba, také viděli zapojení mitochondriální DNA a některých jejích mutací.